










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO Compartir

 Permalink
PermalinkGaceta Médica Boliviana
versión impresa ISSN 1012-2966versión On-line ISSN 2227-3662
Gac Med Bol v.35 n.1 Cochabamba 2012
Caso Clínico
Estudio imagenológico de poliquistosis renal autosómica dominante, reporte de un caso y revisión de la literatura
Imaging study autosomal dominant polycystic kidney disease, report of a case and review of the literature
Sindy Vanessa Panozo Borda1,a, Klonddy Heredia Moy1,a, Ifigenia Oviedo Gamboa1,a, Tania Villarroel Arze1,b, William Zegarra Santiesteban1,c, Rodolfo Ricaldez Muñoz1,b
1Servicio de radiología, Hospital Obrero N° 2 de la Caja Nacional de Salud, Cochabamba, Bolivia.
aResidente de radiología; bMédico radiológo; Jefe del departamente de radiología.
*Correspodencia a: Sindy Vanessa Panozo Borda.
Correo electrónico: vanessa_shg@hotmail.com
Recibido el 4 de octubre de 2011.
Aceptado el 14 de mayo de 2012
Resumen
La poliquistosis renal autosómica dominante (PRAD), es una enfermedad hereditaria multiorgánica, caracterizada por el progresivo crecimiento y desarrollo de quistes renales que destruyen el parénquima funcional. Es la patología quística renal más frecuentemente transmitida de forma genética y es causa de insuficiencia renal crónica (IRC) que en ocasiones precisa de tratamiento renal sustitutivo. Describimos el caso de una paciente adulta con PRAD asociada a poliquistosis hepática que tiene antecedente del progenitor de PRAD, fue diagnosticada hace ocho años por estudio ecográfico, se le realizó el seguimiento correspondiente. Actualmente empezó a presentar alteración de la función renal, pero preserva la función hepática. Existen muy pocos casos reportados en nuestro medio, a pesar de ser una patología relativamente frecuente. Por lo que se decide hacer una revisión enfocada en el diagnóstico imagenológico, dejando en claro la utilidad de la ecografía en el diagnóstico de poliquistosis renal, ya que es el método de elección en el diagnóstico por imagen, adicionalmente nos permite hacer un seguimiento del caso y confirmar o descartar la frecuente asociación de una poliquistosis en otro órgano (fundamentalmente hepático).
Palabras claves: riñón poliquístico autosómico dominante; poliquistosis hepática; quistes; diagnóstico; ecografía.
Abstract
Autosomal dominant polycystic kidney disease (PRAD) is a multisystem hereditary disease characterized by the progressive growth and development of renal cysts that destroy functional parenchyma. Renal cystic disease is the most common genetic form transmitted and causes of chronic renal failure (CRF) which sometimes requires renal replacement therapy. We describe an adult patient with polycystic liver PRAD associated with the parent who has a history of PRAD, was diagnosed eight years ago by ultrasonography, underwent the follow-up. Today started presenting impaired renal function, but preserved liver function. There are very few cases reported in our area, despite being a relatively common condition. It is decided to review focuses on imaging diagnosis, making clear the usefulness of ultrasonography in the diagnosis of polycystic kidney disease, since it is the method of choice for imaging additionally allows us to monitor the case and confirm or rule out the frequent association of polycystic in another organ (mainly liver).
Keywords: polycystic kidney, autosomal dominant; polycystic liver disease; cysts; diagnosis; ultrasound.
La enfermedad poliquística renal, es una enfermedad hereditaria multiorgánica, caracterizada por el progresivo crecimiento y desarrollo de quistes renales que destruyen el parénquima funcional1. El término queda reservado para dos tipos de enfermedades quísticas hereditarias: la poliquistosis renal autosómica dominante (PRAD), mal llamada de «tipo adulto», y la poliquistosis renal autosómica recesiva (PRAR), también mal llamada de «tipo infantil», porque ambas formas pueden también presentarse en el niño2 y en el adulto3. En este artículo nos referimos con más énfasis a la PRAD.
La característica más importante de esta enfermedad es el desarrollo de quistes en los riñones que conducen, generalmente, a un fallo renal en la vida adulta4. En varias ocasiones se asocia a manifestaciones extrarrenales5 o malformaciones asociadas, que incluyen: 1) quistes hepáticos (30 a 60%), 2) quistes pancreáticos (10%), 3) quiste esplénicos (5%), quistes en tiroides, ovario, endometrio, vesículas seminales, pulmón, cerebro, glándula hipófisis, mama y epidídimo, 4) aneurismas cerebrales en baya (18 al 40%)6. Estos representan una causa significativa de morbimortalidad debida a hemorragia subaracnoidea7, 5) aneurisma de la aorta abdominal, 6) lesiones cardíacas y 7) divertículos colónicos6. Sin embargo, la principal causa de morbilidad es la enfermedad renal progresiva caracterizada por la formación y aumento de tamaño de quistes. Debido a esto, la función renal se deteriora y conduce a una enfermedad terminal en más del 50% de los pacientes a la edad de 60 años8.
El diagnóstico precoz de la PRAD conllevaría un mejor pronóstico, al permitir un seguimiento clínico más estricto9, cumpliendo los objetivos terapéuticos de forma temprana y de esa forma retrasar el avance de la enfermedad renal. La hipertensión y la infección renal se deben combatir de manera intensiva apenas se tenga un diagnóstico para preservar el funcionamiento renal10.
La ecografía abdominal es la mejor modalidad de imagen, que se dispone tanto para el diagnóstico, como para hacer el cribado en familias de pacientes afectados, así como en el seguimiento sistemático de pacientes que tienen enfermedad conocida6.
A continuación presentamos el caso de una paciente, que presentó poliquistosis renal asociado a quistes hepáticos, con alteración de la función renal y preservación de su función hepática. Existen muy pocos casos reportados en nuestro medio a pesar de ser una patología relativamente frecuente, por lo que se decide hacer una revisión enfocada en el diagnóstico imagenológico, dejando en claro la utilidad de la ecografía en el diagnóstico de poliquistosis renal.
Presentación del caso
Paciente de 56 años de edad, sexo femenino que presenta un cuadro clínico de ocho años de evolución, caracterizado por haber tenido dolor abdominal constante en región epigástrica e hipocondrio derecho asociado, a masa palpable que llega hasta flanco derecho, además el cuadro se acompaña de infecciones del tracto urinario recurrentes y micro hematuria. Al examen físico presenta abdomen con franca hepatomegalia que llega hasta flanco derecho, aproximadamente 8 cm debajo del reborde costal, ligeramente doloroso a la palpación.
Tiene como antecedente familiar relevante, que su progenitor diagnosticado de PRAD, falleció hace quince años por complicaciones de la misma.
Entre sus exámenes complementarios presenta, una primera ecografía hace ocho años donde se visualizan múltiples imágenes ovoideas anecoicas de bordes lisos y regulares, sin contenido endoluminal que producen refuerzo posterior, de diferente tamaño distribuidas en parénquima hepático y renal, con este estudio ecográfico se realiza el diagnóstico de Enfermedad Poliquística hepatorrenal. Con las manifestaciones clínicas más el antecedente familiar se confirma el diagnóstico de PRAD. Se le realiza seguimiento ecográfico a lo largo de estos ocho años, en el que se observa un aumento de tamaño y número de quistes, con franco crecimiento de ambos órganos a expensas de dichas imágenes antes descritas, además de la alteración de la ecoestructura normal de ambos órganos (fig. 1 y 2).
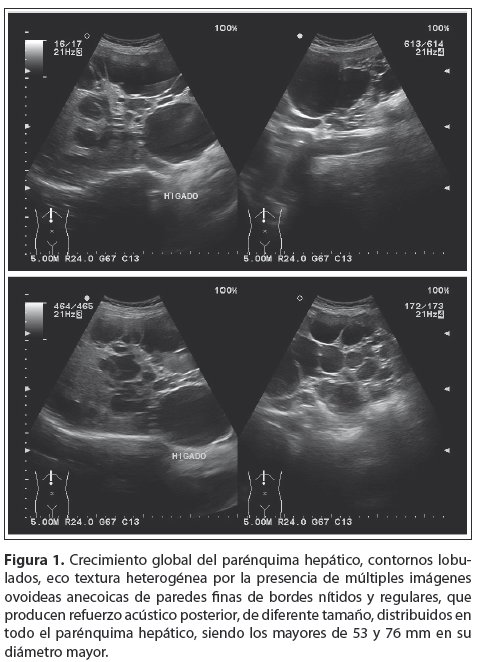
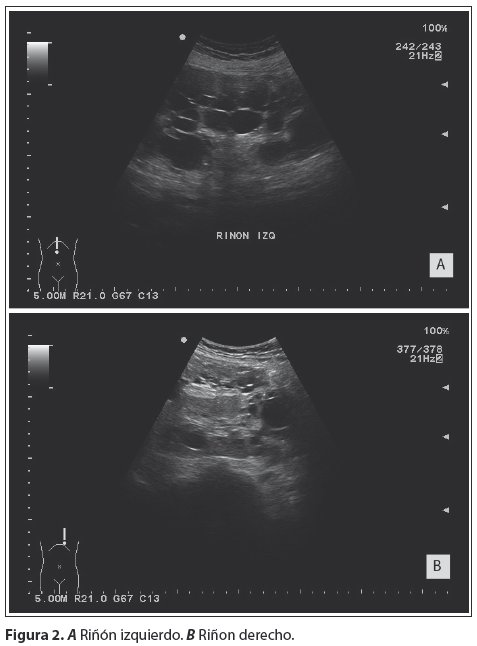
Posteriormente se realiza una Tomografía Computari-zada (TC) con Contraste (fig. 3), que confirma el diagnóstico ecográfico.

Actualmente, después de ocho años del diagnóstico inicial con preservación de la función renal durante todo ese tiempo; la paciente cursa con alteración de la misma, lo que indica nefropatía existente, que concuerda con lo indicado por la literatura consultada, en relación a la edad de la paciente. Sin embargo, se están tomando medidas preventivas para evitar el avance de la enfermedad y sus complicaciones.
Discusión
La poliquistosis renal autosómica dominante (PRAD), es una enfermedad sistémica hereditaria caracterizada por el desarrollo de quistes renales bilaterales11, es la patología quís-tica renal más frecuentemente transmitida de forma genética. Afecta aproximadamente a una de cada 1000 personas y es responsable del 7-10% de los casos de insuficiencia renal crónica (IRC) que precisan tratamiento renal sustitutivo1,12, a diferencia de la PRAR que se presenta en apenas 1 de cada 20000 nacimientos, aproximadamente13.
Suele producir síntomas en el tercer o cuarto decenio, pueden presentar dolor crónico en fosa renal por el efecto de masa del riñón aumentado de tamaño, agudo que indica infección, obstrucción urinaria por un coágulo o cálculo, hemorragia súbita dentro de un quiste y rotura quística. Son frecuentes macrohematuria y microhematuria. La nefrolitiasis afecta al 15 o 20%, la formación de cálculos tiende a ocurrir en pacientes con más quistes y un tamaño del quiste predominante significativamente mayor6. Se detecta hipertensión en 20 a 30% de los niños y hasta en 75% de los adultos, secundaria a isquemia intrarrenal por distorsión de la estructura del órgano y activación del sistema renina-angiotensina. La infección urinaria es frecuente y puede afectar la vejiga, el intersticio renal (pielonefritis) o un quiste (quiste piógeno)10. El desarrollo de quistes en los riñones conduce a un fallo renal en la vida adulta14. Aparece insuficiencia renal en el 50% de los pacientes, y habitualmente ya está presente a los 60 años de edad6.
La manifestación extrarrenal más frecuente es el desarrollo de múltiples quistes hepáticos15. La poliquistosis hepática se caracteriza por el desarrollo gradual de múltiples quistes, posterior al de los quistes renales que son variables en número y tamaño, diseminados por ambos lóbulos del hígado, de contenido seroso, de paredes finas, que no comunican con las vías biliares. Mucho más frecuente en el sexo femenino, los quistes se manifiestan a partir de la pubertad, crecen sobre todo en la época fértil de la mujer y van adquiriendo su máxima expresión a partir de la quinta década de la vida. A pesar de los hallazgos físicos y radiológicos, a veces impresionantes, sólo en una minoría de los pacientes con poliquistosis hepática progresará a lo largo de los años hacia una enfermedad hepática avanzada o desarrollará complicaciones como resultado de hepatomegalia masiva16. Por lo tanto, el pronóstico está determinado por la evolución de la enfermedad renal a insuficiencia renal crónica17,18, ya que los múltiples quistes hepáticos no suelen dar síntomas ni alteran la función hepática.
En el estudio imagenológico, la ecografía es el método de elección, en la que se puede observar riñones grandes con quistes asimétricos bilaterales múltiples de tamaño variable. Los quistes que están complicados por hemorragia o infección muestran paredes gruesas, ecos internos y/o niveles líquido-desechos. Se puede ver calcificación distrófica de la pared de los quistes en forma de focos ecógenos con sombreado acústico distal nítido. Son poco frecuentes los quistes renales en pacientes menores de 30 años. Ravine y cols.19, modificaron los criterios de Bear para el diagnóstico de PRAD y afirman que en los pacientes de 30 años o menos que tienen antecedentes familiares de PRAD son necesarios dos quistes renales (unilaterales o bilaterales) para hacer el diagnóstico. En los pacientes de 30 a 59 años de edad son necesarios dos quistes en los dos riñones, y a los 60 años de edad o más son necesarios cuatro quistes en cada riñón6. La Tomografía Computarizada muestra lesiones de densidad agua, sin pared y que no captan contraste. Ambas técnicas proporcionan información detallada sobre el tamaño, número y localización de los quistes19, así como sobre la existencia de malformaciones asociadas. La Resonancia Magnética posee un alto poder resolutivo, tanto para el diagnóstico como para la detección de complicaciones intraquísticas20.
Por prevalencia y semejanza clínica, la PRAR constituye la principal entidad con la que se plantea el diagnóstico di-ferencial21. Establecer clínicamente la diferencia entre PRAD, mucho más frecuente en la población, y PRAR no siempre es sencillo22, ya que en general se manifiesta demasiado tarde. Entonces es necesario tomar en cuenta los estudios de imagen, el patrón de herencia e inclusive el estudio genético.
Las técnicas de imagen permiten diferenciar la PRAR y la PRAD entre sí22. Se resume en la tabla 1 las características ecográficas, para realizar el diagnóstico diferencial, tanto en niños como en adultos.

Se ha descrito que el tamaño renal en los pacientes con PRAR disminuye progresivamente con la severidad de la IRC23. Asimismo, en adultos con PRAD y filtrado glomerular todavía normal los riñones no están francamente aumentados de tamaño en un porcentaje valorable de casos, y la nefrome-galia se desarrolla posteriormente en el curso evolutivo de la enfermedad22.
Una historia familiar negativa de poliquistosis, la normalidad ecográfica renal en los progenitores e incluso la histología hepática compatible con PRAR permiten el diagnóstico23.
Los objetivos terapéuticos de la PRAD, consisten en retrasar el avance de la enfermedad renal y reducir al mínimo los síntomas. La hipertensión y la infección renal se deben combatir de manera intensiva para preservar el funcionamiento renal. Los inhibidores de la enzima convertidora son antihipertensivos eficaces, pero hay que vigilar de cerca a los pacientes, porque algunos llegan a sufrir insuficiencia renal e hiperpotasemia. Las infecciones urinarias se tratan de manera habitual, salvo que se sospeche un quiste piógeno, en cuyo caso deben utilizarse antibióticos que penetren dentro el quiste. El dolor crónico causado por los quistes se trata mediante punción y esclerosis del quiste con etanol10.
Por lo tanto, es importante recalcar que la ecografía abdominal es el método de elección de imagen, tanto en el diagnóstico de PRAD de pacientes nuevos, como en el cribado de familias de pacientes conocidos, así como en el seguimiento sistemático de pacientes ya diagnosticados. Ya que es un método de imagen accesible, en el cual no se utiliza radiación ionizante y en el que la relación costo beneficio es elevada. Otro de sus beneficios, es que permite ver la probable asociación de malformaciones extrarrenales (poliquistosis hepática la más frecuente) y las posibles complicaciones si es que se presentan. Es así que concluimos que la PRAD es una enfermedad relativamente frecuente, que conlleva a complicaciones muchas veces letales para los pacientes, consideramos que el diagnóstico precoz es imprescindible para un tratamiento oportuno de las complicaciones y así retrasar el avance de la enfermedad renal.
Conflictos de interés: los autores declaran no tener conflictos de interés en la publicación de este caso.
Referencias bibliográficas
1. Bleyer AJ, Hart TC. Polycystic kidney disease. N Engl J Med 2004; 350(25): 2622. [ Links ]
2. Gagnadoux MF, Broyer M. Polycystic kidney disesase in children. En: Davison AM, Cameron JS, Grünfeld J-P, Keer DUS, Ritz E, Winearls CD (Eds). The Oxford Textbook of Clinical Nephrolo-gy on CD-ROM. Chapter 16.2.1, Oxford Univer-sity Press; 1997.
3. Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol 1997; 11(3): 302-6. [ Links ]
4. Torra R, Badenas C, Darnell A, Nicolau C, Vol-pini V, Revert L, et al. Estudio clínico, genético y molecular de la poliquistosis renal autosómica dominante tipos 1 y 2. Med Clin (Barc) 1998; 110: 481-7. [ Links ]
5. Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med 1993; 329(5): 33242. [ Links ]
6. Rumack CM, Wilson SR, Charboneau JW, Jo-hnson J-AM. Diagnóstico por ecografía. 3a ed. Madrid: Elsevier; 2006; p.372-373.
7. Parfrey PS, Bear JC, Morgan J, Cramer BC, McManamon PJ, Gault MH, et al. The diagnosis and prognosis of autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323: 1085-90. [ Links ]
8. Asplin JR, Coe FL. Enfermedades tubulares hereditarias. En: Fauci AS, Braunwald E, Isselbacher K, Wilson J, Martín J, Kasper D, et al. Harrison Principios de Medicina Interna. 14 ed. Madrid: McGraw Hill, 1998.
9. Sujansky E, Kreutzer SB, Johnson AM, Lezotte DC, Schrier RW, Gabow PA. Attitudes of at-risk and affected individuals regarding presymptoma-tic testing for autosomal dominant polycystic kid-ney disease. Am J Med Genet 1990; 35(4): 510-5. [ Links ]
10. Harrison, Kasper DL, Braunwald E. Principios de medicina interna. 16 ed. México: McGraw-Hill Interamericana; 2006. p. 1868-1870. [ Links ]
11. Peces R, Venegas J. Quistes de las vesículas semifinales e infertilidad en la poliquistosis renal autosómica dominante. Nefrologia 2005; 25: 7880. [ Links ]
12. Grantham JJ. The etiology, pathogenesis, and treatment of autosomal dominant polycystic kid-ney disease: recent advances. Am J Kidney Dis 1996; 28(6): 788-803. [ Links ]
13. Zerres K, Macher G, Becker J. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical ex-perience, and fetal morphology. Am J Med Genet 1998; 76: 137-144. [ Links ]
14. Torra R, Badenas C, Darnell A, Nicolau C, Volpini V, Revert L, et al. Estudio clínico, genético y molecular de la poliquistosis renal autosómica dominante tipos 1 y 2. Med Clin (Barc) 1998; 110: 481-7. [ Links ]
15. Hansmann MF, Ryan JA, Holmes JH, Hogan S, Lee FT, Kramer D,et al. Management and long term follow up of hepatic cysts. Am J Surg 2001; 181: 404-10. [ Links ]
16. García-Gil F, Güemes Sánchez A, Esteban Grau E, Lamata Hernández F, Sousa Domínguez
R, Serrano Aulló M. Trasplante de hígado en la poliquistosis hepática gigante con insuficiencia hepática terminal. Rev Esp Enferm Dig 2008; 100(1): 59-60.
17. Nagorney DM. Surgical management of cystic disease of the liver. En: Blumgart LH, Fong Y. Sur-gery of the liver and biliary tract. 3rd ed. London: W.B. Saunders; 2000.; p. 1261-76. [ Links ]
18. Parks RW, Garden OJ. Benign liver lesions. En: Garden OJ. Hepatobiliary and pancreatic surgery. London: W.b. Saunders Company; 2001; p. 75105. [ Links ]
19. Ravine D, Walker RG, Gibson RN, Sheffield LJ, Kincaid-Smith P, Danks DM. Treatable com-plications in undiagnosed cases of autosomal do-minant polycystic kidney disease. Lancet. 1991;337(8734): 127-9. [ Links ]
20. De Almeida E, Martins Prata M, de Almeida S, Lavinha J. Long-term follow-up of a family with autosomal dominant polycystic kidney disease type 3. Nephrol Dial Transplant 1999; 14(3): 6314. [ Links ]
21. Murcia NS, Woychik RP, Avner ED. The molecular biology of polycystic kidney disease. Pediatr Nephrol 1998; 12(9): 721-6. [ Links ]
22. Ariceta G, Lens X. Poliquistosis renal autonómica recesiva. Nefrologia. 2003; 23(5): 23-7. [ Links ]
23 Zerres K, Rudnik-Schoneborn S, Steinkamm C, Mucher G. Autosomal recessive polycystic kidney disease. Nephrol Dial Transplant 1996; 11 Suppl 6: 29-33

