GLUCOGENOSIS: CARACTERIZACIÓN CLÍNICA - PATOLÓGICA DEL PRIMER CASO DESCRITO EN BOLIVIA EN UN PACIENTE PEDIÁTRICO
GLYCOGEN STORAGE DISEASE: FIRST CLINICOPATHOLOGIC PEDIATRIC CASE DESCRIBED IN BOLIVIA
Dr. MSc. Juan Pablo Rodríguez Auad1, Dra. Rita Villalobos2, Dra. Beatriz Luna3, Dr. Luis Tamayo4
1 Pediatra Infectólogo. Máster en Ciencias Médicas. Hospital del Niño "Ovidio Aliaga Uría". juparodau@hotmail.com. Cel 60568970
2 Anatomopatóloga. Hospital del Niño "Ovidio Aliaga Uría". ]]>
RECIBIDO: 29/09/15
ACEPTADO: 07/10/15
RESUMEN
Las glucogenosis son enfermedades hereditarias del metabolismo del glucógeno. Se reconocen más de 12 tipos y afectan principalmente al hígado y al músculo, mismas que se clasifican según la enzima defectuosa y el órgano afectado.
Presentamos el caso de un niño de 4 años y 6 meses con hepatomegalia importante, retardo del crecimiento pondoestatural, extremidades delgadas, facies con mejillas redondas. Sus exámenes laboratoriales revelaron: hipoglicemia, hiperlipidemia, hiperuricemia y sus estudios imagenológicos evidenciaron hepatomegalia difusa severa. El estudio histopatológico concluyó con glucogenosis, no pudiendo definirse el tipo, por la imposibilidad de realizar pruebas específicas de histoquímica en Bolivia. El paciente es seguido por consulta externa, bajo indicaciones dietéticas para prevenir complicaciones.
Palabras clave: glucogenosis, enfermedad de Von Gierke.
Glycogen storage diseases are inherited metabolic disorders of glycogen metabolism. There are over 12 types, they may affect primarily the liver and muscle. They are classified and the affected tissue. The case of a 4 y 6m old-male infant is presented, with growth retardation, thin limbs, rounded cheeks. Laboratory testing showed hypoglycemia, hyperlipidermia, hyperuricemia. Imagenoly testing showed severe diffuse hepatomegaly. Histopathology concluded in glycogen storage disease, the enzyme deficiency could not be established because of the unavailability of these test in Bolivia. The patient is followed by consult, diet therapy to prevent complications.
Key words: glycogen storage disease, Von Gierke's disease.
INTRODUCCIÓN
Los trastornos genéticos del metabolismo son enfermedades causadas por mutaciones en genes individuales que codifican proteínas específicas. Estas mutaciones pueden traducirse en alteraciones de la estructura de la proteína sintetizada y su función.
Muchas de estas mutaciones no tienen consecuencias clínicas y representan diferencias polimorfas que sirven para diferenciar a las personas, pero algunas mutaciones ocasionan enfermedades leves e incluso mortales. Los hidratos de carbono más importantes son los monosacáridos (glucosa, galactosa y fructosa) y un polisacárido (glucógeno). El glucógeno es la principal fuente de energía almacenada en el músculo.
La glucosa es el principal sustrato del metabolismo energético y sus concentraciones sanguíneas son mantenidas con dieta, gluconeogénesis y glucogenólisis.1,2
En la glucogenosis se produce el depósito de glucógeno en los tejidos encargados del metabolismo del glucógeno como el hígado y el músculo, por deficiencia genética de la actividad de alguna de las enzimas que degrada o sintetiza el glucógeno2.
]]> Las manifestaciones clínicas de la glucogenosis expresan la dificultad que existe en estos tejidos para movilizar sus depósitos de glucógeno; si el hígado es afectado, se produce hepatomegalia, alteraciones en la glucemia y falla en el crecimiento. Si el músculo está afectado suele manifestarse con debilidad muscular, fatiga y dolor muscular entre otros3. La frecuencia de todas las formas de glucogenosis es de aproximadamente 1/20.000 a 1/25.000 nacidos vivos4,5 y padres con un niño con glucogenosis tienen un 25% de posibilidad de tener otro hijo con glucogenosis.Existen más de 12 formas de glucogenosis, se clasifican según el órgano afectado en glucogenosis hepáticas y musculares. Los déficit de glucosa-6-fosfatasa (tipo I), glucosidasa ácida lisosómica (tipo II), enzima desramificadora (tipo III) y fosforilasacinasa hepática (tipo IX) son las más frecuentes en la infancia. Las glucogenosis que afectan principalmente al hígado incluyen los déficits de glucosa-6-fosfatasa (tipo I) y enzima desramificadora (tipo III). La glucogenosis suelen cursar con hipoglucemia en ayunas y hepatomegalia. Otros órganos también pueden estar afectados y pueden manifestarse como disfunción renal en el tipo I, o miopatía (esquelética y/o miocárdica) en el tipo III y IV5.
CASO CLÍNICO
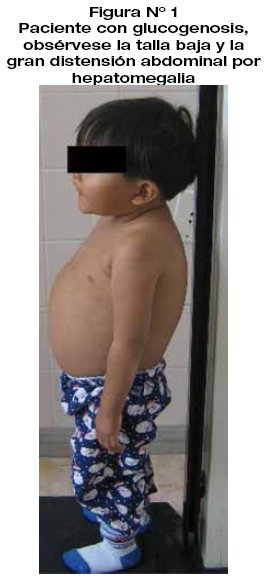
Niño de 4 años y 6 meses de edad, procedente y residente del municipio de San Antonio de Los Yungas (La Paz, Bolivia). Madre de 18 años, padre de 27 años, hermano de 3 años aparentemente sanos. Nacido por cesárea con un peso al nacimiento de 3.6 Kg y una talla de 49cm. A la edad de 1 año se observa aumento del perímetro abdominal e inadecuado crecimiento motivo por el cual fue internado en hospital de segundo nivelpor hepatomegalia en estudio siendo egresado sin diagnóstico etiológico. En febrero de 2014 se interna en un hospital privado de la ciudad de La Paz donde se identifica Schistosoma japonicum en coproparasitológico y al cual se atribuye la hepatomegalia. Posteriormente es transferido al Hospital del Niño Dr. Ovidio Aliaga Uría, de La Paz al servicio de Infectología con el diagnóstico de hepatomegalia en estudio y probable esquistosomiasis. El examen físico reveló un peso de 14 Kg, talla de 84 cm,(-3 DS, con talla baja) perímetro cefálico de 49cm, signos vitales estables, marcada distensión abdominal a expensas de hepatomegalia severa (llegaba hasta fosa iliaca derecha a 10 cm del borde costal). (Fig. 1)
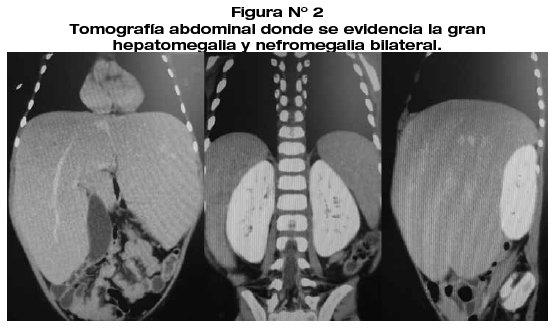
Por el tiempo de evolución del cuadro clínico y las buenas condiciones generales del paciente (ausencia de fiebre, ausencia de síndrome de respuesta inflamatoria sistémica y buena tolerancia oral) se descartó la posibilidad de proceso infeccioso y/o neoplásico por lo que se sospechó en enfermedad por depósito o enfermedad metabólica a nivel hepático. Los estudios de laboratorio concluyeron en: glucosa sérica en ayuno: 47 mg/ dL, ácido úrico: 4.3 mg/dL, Colesterol: 370 mg/dL y Triglicéridos: 240 mg/ dl. Los estudios de imagenología revelaron hepatomegalia difusa y nefromegalia bilateral.(Fig.2).
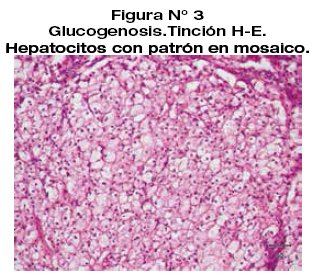
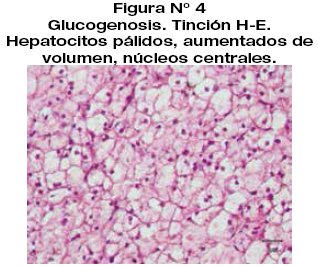
Se realiza examen de inmuno histoquímica extra institucional para CD68 y CD163 que fueron negativos. La tinción para citoqueratina y AHE es positiva en las células antes descritas. Hallazgos histológicos e inmunohistoquímicos compatibles con Glucogenosis.
DISCUSIÓN
Para el diagnóstico de glucogenosis es necesaria una adecuada anamnesis con búsqueda intencionada de otros probables familiares afectados, así como la duración de los síntomas. El examen físico es importante, la antropometría, ya que estos pacientes tienen compromiso en su crecimiento, sobre todo talla baja.
La hepatomegalia es un hallazgo muy frecuente, nuestro paciente presentaba una hepatomegalia severa. Se ha descrito que estos niños suelen tener una "cara similar a la de las muñecas", con mejillas redondeadas, extremidades delgadas, baja estatura y abdomen protuberante. Nuestro paciente presentaba todas estas características. Las pruebas de laboratorio evidenciaron hipoglucemia e hiperlipidemia, pruebas que pueden orientar al diagnóstico. La determinación de glucosa sérica en ayuno es fundamental ya que muchas de estas glucogenosis cursan con hipoglucemia en ayuno. Como una manera de compensar el deficiente metabolismo hepático de la glucosa existe también en estos pacientes elevación del ácido láctico. Otras pruebas que también se deben solicitar son pruebas de función hepática y enzimas musculares (creatinfosfocinasa=CPK). En las pruebas de función hepática las transaminasas y bilirrubinas pueden estar normales o levemente aumentadas como se observó en nuestro paciente, las cuales no concuerdan con la hepatomegalia con la que cursan frecuentemente estos pacientes. También se debe solicitar ácido úrico y perfil de lípidos ya que estos pacientes presentan hiperuricemia, hipercolesterolemia e hipertrigliceridemia. En el paciente en estudio sólo se observó elevación de los lípidos y no así del ácido úrico. Se observó el aspecto "lechoso" del plasma en nuestro paciente debido a la hiperlipidemia. El hemograma, plaquetas y tiempo de coagulación suelen estar normales, aunque en algunas formas de glucogenosis existe afectación hematológica con leucopenia. En general nuestro paciente no tuvo afectación hematológica y la eosinofilia que presentó se atribuyó a la enteroparasitosis que fue tratada sin complicaciones. Otros estudios de laboratorio que se recomiendan, pero que no se realizaron en el paciente, son pruebas de sobrecarga oral de glucosa.
]]> Los estudios de imagenología permiten determinar el grado de afectación del hígado. La ecografía, la tomografía axial computarizada y/o la resonancia magnética abdominal son estudiosútiles, no sólo para ver la magnitud de las visceromegalias, sino para el diagnóstico diferencial con tumores, quistes, malformaciones. Uno de los principales estudios diagnósticos en casos de glucogenosis en nuestro medio y en muchas partes del mundo es la biopsia hepática, la cual permite confirmar el depósito de glucógeno hepático intrahepatocitario, evaluar la presencia de grasa, fibrosis, cirrosis u otras alteraciones. El estudio de histoquímica confirma el diagnóstico con la determinación del nivel de actividad de la enzima afectada en el hígado, en el caso de la glucogenosis tipo I, se deberá medir el nivel de actividad de la glucosa-6-fosfatasa. El análisis de las mutaciones de los genes implicados ofrece un método de diagnóstico no invasivo y preciso. Sin embargo estos últimos estudios de histoquímica y genética son de elevado costo y no están disponibles en muchos países, incluyendo el nuestro. En el caso descrito, se llegó al diagnóstico de glucogenosis por las características clínicas, estudios de laboratorio e imagen y características de la biopsia hepática.No existe un tratamiento específico para la glucogenosis, sin embargo es fundamental un adecuado tratamiento dietético para prevenir y reducir la posibles complicaciones: hiperlipidemia, pancreatitis, arteriosclerosis, adenomas hepáticos, afectación renal (nefromegalia bilateral, proteinuria, hipertensión arterial, litiasis renal, nef rocalcinosis, glomeruloesclerosis), hiperuricemia (episodios de gota), osteoporosis y cirrosis hepática; se menciona el trasplante hepático en algunos casos5. Se han reportado alteraciones endocrinológicas en algunos pacientes.6 Se debe prevenir la hipoglucemia y sus consecuencias metabólicas, con comidas frecuentes y ricas en hidratos de carbono o la administración de almidón crudo de maíz (maicena). Se sugiere que el contenido de la dieta siga la siguiente distribución: hidratos de carbono 60-65%; proteínas 10-15% y lípidos 20-30%del aporte calórico total. En casos donde la hipoglucemia es frecuente, se debe administrar nutrición enteral nocturna por medio de infusión de glucosa a través de una sonda nasogástrica. Nuestro paciente presentó hipoglucemia en ayuno, sin embargo sus controles de glucosa sérica durante el día fueron normales antes y posterior a la alimentación; en ningún momento hubo datos de hipoglucemia sintomática. Sin embargo se hizo una valoración nutricional y fue egresado con un régimen nutricional rico en hidratos de carbono de absorción lenta que incluía al almidón de maíz con restricción de aquellos hidratos de carbono que no se pueden convertir directamente en glucosa (fructosa, galactosa, sucrosa, lactosa). Debido a estas restricciones dietéticas, las vitaminas y los minerales como el calcio y la vitamina D pueden ser deficitarios, por lo que es necesario suplementar a estos pacientes con vitaminas yminerales, especialmente con calcio, para cubrir los requerimientos. El tratamiento dietético mejora la hiperuricemia7, la hiperlipidemia y la función renal, retrasando el desarrollo de la insuficiencia renal. Para el control de la hiperuricemia se puede usar alopurinol8, en el caso presentado no fue necesario su uso debido a que no se evidenció incremento del ácido úrico, sin embargo se requerirán controles posteriores y seguimiento laboratorial metabólico. El paciente fue egresado en buenas condiciones generales, se dio una explicación detallada a la madre sobre la enfermedad, cuidados nutricionales y dietéticos, complicaciones y seguimiento estricto por consulta externa. El manejo del paciente fue multidiscliplinario (Pediatría, Gastroenterología, Nutrición, Genética, Nefrología e Infectología). Finalmente, dada la imposibilidad de hacer un diagnóstico enzimático y genético en nuestro medio, no se pudo definir el tipo de glucogenosis, sin embargo por las características clínicas, laboratoriales y biopsia se sostiene una glucogenosis de tipo I (déficit de glucosa-6 fosfatasa o translocasa) llamada también Enfermedad de Von Gierke9,10. La glucogenosis tipo I es una enfermedad autosómica recesiva, con mutaciones genéticas conocidas y es posible realizar la detección de portadores mediante pruebas de diagnóstico basadas en ADN (ácido desoxirribonucleico)11,12.
Existen 4 subtipos dependiendo de la anomalía en el sistema de la glucosa-6-fosfatasa: la subunidad catalítica del sistema se encuentra en el retículo endoplásmico y su defecto causa la glucogenosistipo Ia, adicionalmente existen transportadores para la entrada del sustrato glucosa-6-fosfato al retículo endoplásmico, el defecto del transportador es el tipo Ib, adicionalmente se ha postulado la existencia del tipo Ic, y el Id.13
En nuestro paciente se tomó muestra de sangre en tubo con EDTA y se extrajo el ADN de esta muestra para posibles estudios genéticos futuros.
El pronóstico de estos pacientes es reservado, aunque muchos pueden vivir hasta la edad adulta, conforme crece el paciente las complicaciones metabólicas, hepáticas, renales, óseas, vasculares, etc., pueden ser más frecuentes por lo que un adecuado seguimiento y control de estas complicaciones mejorará su calidad de vida. El diagnóstico precoz y el tratamiento adecuado desde la infancia mejoran el pronóstico y la sobrevida a largo plazo de estos pacientes. Si bien las glucogenosis son poco frecuentes muchas veces no son diagnosticadas e inclusive muchos casos se diagnostican en autopsias14,15. Es por eso que el conocimiento de estas enfermedades permitirá al clínico buscarlas dentro de los diagnósticos diferenciales de niños con hepatomegalia.
REFERENCIAS
1. Fernandez-Teijeiro A. Masas abdominales. En: Arguelles M F, Arguelles A F, Eds, Urgencias en gastroenterología, hepatología, y nutrición pediátrica. Madrid: Ergon. 2011. p. 233-42. [ Links ]
2. Mc Bride K. Enfermedad de Wilson. En: Bishop W, Ed, Gastroenterología Pediátrica práctica. Venezuela: Amolca 2012.p. 356-66. [ Links ]
3. Mahler R.Glycogen storage disease. J. Clin. Path.,22, suppl.2014; 2, 32-41.
]]>4. Rake JP,Visser G, Labrune Ph, Leonard J, Ullrich K, Smit P. Guidelines for management of glycogen storage disease type I. European Study on Glycogen Storage Disease Type I .Eur J Pediatr. 2002; 161; 112-119. [ Links ]
5. Alvear C, Barboza M, Rodríguez ZK. Glycogen storage disease: report of two cases in the city of Cartagena. Colomb. Med.2010;41:76-81. [ Links ]
6. Cornejo V, Raimann E.Glucogenosis tipo I y III. Rev Chil Nutr.2006;33;2:135-141. [ Links ]
7. Melis et al.: Involvement of endocrine system in a patient affected by Glycogen storage disease 1b: speculation on the role of autoimmunity. Ital J Pediatr. 2014; 40: 30 [ Links ]
8. Leon Bojorge RC, Belmont-Martinez B. Enfermedad de Pompe forma infantil (glucogenosis tipo II). Informe de dos casos en niños mexicanos descubiertos por autopsia. Acta Pediatr Mex. 2009;30:142-7. [ Links ]
9. Parker P, Ballew M, Greene H. Nutritional Management of Glycogen Storage Disease. Annu Rev Nutr. 1993;13:83-109. [ Links ]
10. Carvalho P, Marques N, Dinis P, Cordeiro J, Conceicao L. Nascimento J. Glycogen Storage Disease type 1a-a secondary cause for hyperlipidemia: report of five cases. J Diabetes MetabDisord. 2013; 12: 25-31. [ Links ]
11. Boers S, Visser G, Smit P, Fuchs SA. Liver transplantation in glycogen storage diseasetype I. Orphanet Journal of Rare Diseases. 2014;9:47. [ Links ]
12. Manzia T, Angelico R, Toti L, Cillis A,Ciano C, Orlando G, et al.Glycogen Storage Disease Type Ia and VI Associated with Hepatocellular Carcinoma: Two Case Reports.Transplant Proc. 2011 May;43(4):1181-3. [ Links ]
13. Bayraktar Y, Özen O. Glycogen storage diseases: New perspectives. World J Gastroenterol. 2007;14:2541-53. [ Links ]
14. Gholami S, Sadr-Nabavi A. Genetic and Glycogen Storage Diseases. J Res Med Sci. 2013; 15(10): 7-11. [ Links ]
15. Özen H. Glycogen storage diseases: New perspectives. World J Gastroenterol.2007; 13(18): 2541-53. [ Links ]
]]>