Síndrome de Hurler Scheie, a propósito de un caso
Hurler Schie syndrome, a case report
Dr.: Jorge Barriga Oropeza*, Consuelo Murillo Sánchez*, Julio Agreda Guerrero*, Omar Andia Alacoria**
* Médico Pediatra. Hospital Daniel Bracamonte. Potosí
** Médico Residente de Pediatría. Hospital Daniel Bracamonte. Potosí
Resumen
Presentamos el caso de un niño de ocho años de edad, que llega al Hospital Daniel Bracamonte de la ciudad de Potosí procedente de área rural consultando por presentar deterioro del sentido de la vista en forma progresiva, al igual que otros síntomas concomitantes. Realizado el examen físico y exámenes complementarios, se llega a la conclusión diagnóstica de Síndrome de Hurler Scheie, que es una mucoplisacaridosis rara en nuestro medio. Aprovechamos el caso para hacer una descripción de esta enfermedad.
Palabras Claves:
Rev Soc Bol Ped 2011; 50 (1): 10-2: Síndrome de Hurler Scheie, mucoplisacaridosis.
Abstract:
We present the case of an eight-year-old boy, admitted into "Daniel Bracamonte Hospital" in the city of Potosi. He carne from a rural area and showed a progressive impairment of visión, as well as other related symptoms. Once the physical and complementary tests were performed, it was concluded that he suffered from Hurler Scheie's syndrome, which is a mucopolysaccharidosis, rare in our environment. We took this case to describe the disease.
Key words:
]]> Rev Soc Bol Ped 2011; 50 (1): 10-2: Hurler Scheie Syndrome, mucopolysaccharidosis.
Introducción
Las mucopolisacaridosis (MPS) son un grupo de enfermedades metabólicas hereditarias causadas por la ausencia o el malfuncionamiento de ciertas enzimas necesarias para el procesamiento de moléculas llamadas glicosaminoglicanos o glucosaminoglucanos, que son cadenas largas de hidratos de carbono presentes en cada una de nuestras células que ayudan a construir los huesos, cartílagos, tendones, córneas, la piel y el tejido conectivo. Lo glicosaminoglicanos (antes llamados mucopolisacaridos) también se encuentran presentes en el liquido que lubrica las articulaciones.
En general los niños que padecen algún tipo de mucopolisacaridosis no producen suficiente cantidad de una de las 11 enzimas requeridas para trasformar estas cadenas de glucosa en proteínas y moléculas más sencillas, o bien producen enzimas que no funcionan correctamente. Los glucosaminoglicanos al pasar el tiempo se acumulan en las células, la sangre y el tejido conectivo. Esto produce daños tisulares permanentes y progresivos que afectan el aspecto y las capacidades físicas, los órganos y también el desarrollo mental.
El Síndrome de Hurler Scheie forma parte de este grupo de enfermedades metabólicas hereditarias que se las denomina genéricamente como mucopolisacaridosis. En la oportunidad presentamos el caso clínico de un niño con esta patología poco frecuente en nuestro medio.
Caso clínico
Niño de 8 años de edad, que fue atendido en el Hospital Daniel Bracamonte de Potosí. Procedente del área rural, con el antecedente de presentar pérdida progresiva de la visión, que le dificulta la lectura y el desenvolvimiento en sus actividades habituales. También informa que presenta cefalalgia occipital que se exacerba con la lectura, pérdida progresiva de la capacidad visual a distancia que le dificulta la visión y desenvolvimiento en la escuela. No tiene antecedentes perinatales ni familiares de importancia.
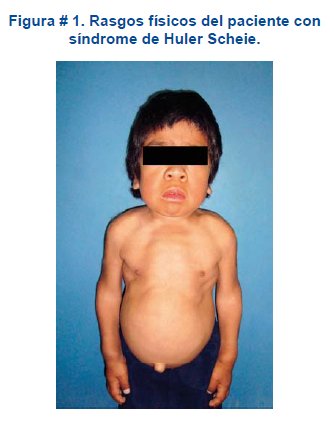
]]> Al examen físico niño con buen estado general orientado en tiempo y espacio. Peso: 17 Kg, talla: 97 cm y termodinámicamente estable. Cráneo ligeramente dolicocéfalo, cabellos negros y abundantes. Facies tosca, ojos grandes ligero exoftalmos con reflejos consensual, fotomotor y de acomodación conservados, opacidad corneal. Boca con labios superior e inferior gruesos, pronunciados. Lengua húmeda rosada con ligera protrusión. Tórax en quilla. Pulmones sin particularidades llamativas. En corazón se percibe un soplo pan diastólico en foco mitral. Abdomen globoso con sobre levantamiento en la línea media xifopubiana atribuible a una diastasis de los músculos rectos anteriores. Genitales acordes con la edad sin particularidad alguna. Extremidades con dificultad para la extensión completa por anquilosis parcial de las articulaciones del codo y rodilla. Dedos de las manos y de los pies con dificultad en la flexión y extensión completa por limitación de los movimientos a nivel de las articulaciones, fuerza muscular disminuida, sensibilidad conservada. Ver figura # 1.
Se realizaron estudios complementarios, dentro de los cuales resaltan: hormonas tiroideas normales, química sanguínea normal, hemograma completo normal, examen general de orina dentro de parámetros normales. Radiografia de tórax: con arcos costales no acordes con la edad cronológica, cartílagos costales osificados. Radiografñia de cráneo que muestra un dolicocéfalo, suturas craneales completamente osificadas y radiografía de los huesos largos de los miembros que evidencian un desarrollo prominente de los extremos epifisiarios.
Con todos los datos clínicos y radiológicos, se llego a la conclusión de que se trataba de una mucoplisacaridosis y probable síndrome de Hurler Scheie.
Discusión
El Síndrome de Hurler tiene una incidencia de acuerdo a diferentes estudios de 1 por 100.000 nacidos vivos y las mucopolisacaridosis se presentan con frecuencia de 1 por 25.000 nacimientos.
Los síntomas del síndrome de Hurler-Scheie generalmente aparecen entre los 3 a 8 años de edad, como el caso de nuestro paciente: Los niños al nacer parecen normales y la enfermedad se caracteriza por un deterioro progresivo, hepatoesplenomegalia, enanismo y cara característica (facies de gárgola) razón por la cual la enfermedad también se denomina gargolismo: rasgos faciales gruesos y toscos con puente nasal deprimido y frente abultada. El sentido de la vista se halla comprometido por que se presenta opacidad corneal, degeneración retiniana, glaucoma. La audición esta disminuida. También es característica la hipertricosis generalizada. En algunos casos se presentan síntomas cardiológicos originados por un prolapso de la válvula mitral.
Los niños al nacer pueden tener mayor peso que el normal, presentando algunas veces hernias inguinal o umbilical. El crecimiento al principio es normal pero luego va disminuyendo finalizando alrededor de los 3 años, presentando entonces un tronco corto. Existe viceromegalia, se presentan deformidades óseas: lordosis lumbar con costillas en forma de remo. Los niños con este síndrome presentan retraso mental progresivo que es un rasgo común en las formas graves y en casi todas las mucopolisacaridosis. Estos niños mueren generalmente alrededor de los 10 años por enfermedad obstructiva de las vías respiratorias, infecciones respiratorias recurrentes o complicaciones cardiacas.
]]> El diagnóstico se efectúa por la clínica y por exámenes de laboratorio especiales: mucopolisacaridos en orina. Otras pruebas incluyen las genéticas para genes que determinar el tipo de síndrome específico. El diagnostico prenatal se lo realiza mediante amniocentesis y estudio de las vellosidades corionicas, para verificar si el feto lleva una copia defectuosa del gen o se encuentra afectado por la enfermedad.Las mucopolisacaridosis, presentan características que se expresan en signos radiológicos agrupados en la llamada disostosis múltiple, así los principales de estos signos serian: cráneo grande, alargado y bóveda craneana engrosada; silla turca en forma de zueco o bota; cuerpos vertebrales hipoplasicos en áreas antero superiores y apariencia de pico; costillas engrosadas con excepción de la inserción espinal en forma de "remo"; metacarpianos co apariencia de "biberón"; humero y cubito con angulación distal; pelvis con proyección del hueso iliaco, acetábulo poco profundo y coxa valga progresiva y huesos largos de aspecto corto y gruesos.
El pronóstico es reservado y esta de acuerdo al grado o tipo de mucopolisacaridosis y depende de las posibles complicaciones cardio-respiratorias.
Las terapias de reemplazo enzimático están actualmente en uso, como la iarunidasa que es útil en la reducción del dolor y disminución de los síntomas neurológicos. Los trasplantes de medula ósea y de sangre del cordón umbilical, pueden utilizarse para el tratamiento del las mucopolisacaridosis, mejorando las características física a excepción de las que afectan el esqueleto y los ojos; pero son procedimientos de alto riesgo con altas tasas de morbilidad.
Referencias
1. Arando D. Síndrome de Hurler, a propósito de un caso. Rev Chil Dermatol 2001 ;23:143-6. [ Links ]
2. Behrman, MD. Richard E. Kliegman, MD. Robert M., Jonson MD. Hald B. Nelson Tratado de Pediatría. Mucopolisacaridosis. 17° edición. España. Elsevier. 2006.p.482-6.
3. Guerrero Fernandez J., Ruiz Domínguez J. A., Menéndez Suso J. J., Barrios Tascon. Manual de Diagnóstico y Terapéutica en Pediatría. 5ta edición - Barcelona - Publimet. Año 2009; 168-169. [ Links ]
]]>