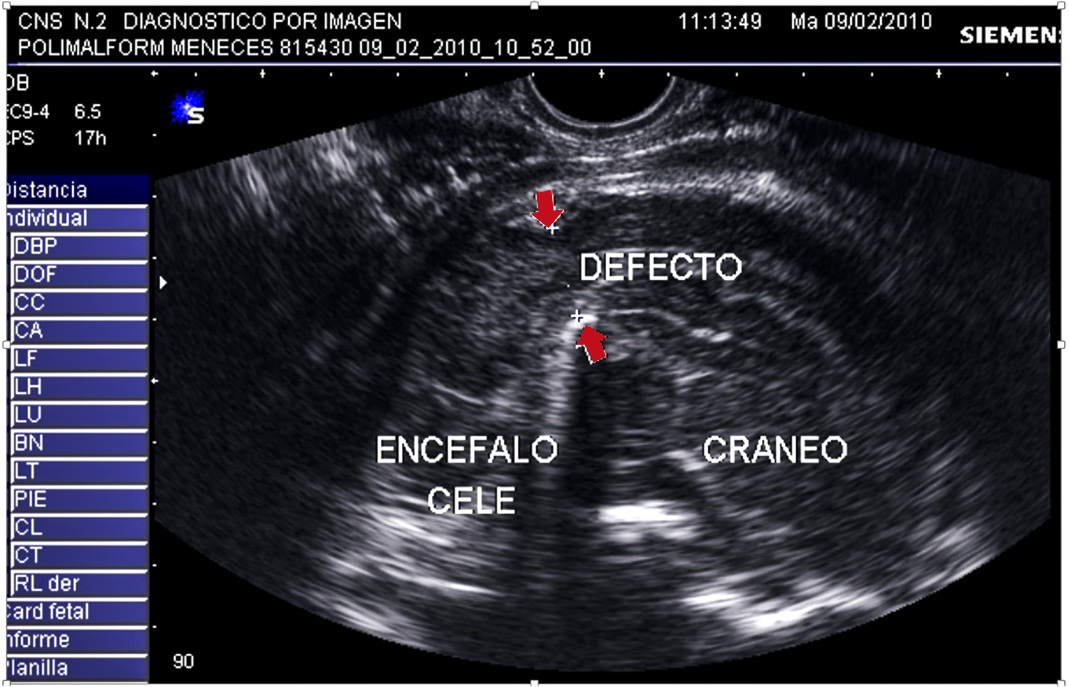
Figura 1:Se muestra el defecto en el hueso occipital por donde se hernia el encéfalo y sus cubiertas.
Maita Q Freddy1,a, Sánchez Soria Galvarro Karla1, Hochstatter Erwin1,b, Camata Magalic.
1Ginecólogo Obstetra.
aUnidad de Medicina fetal, Servicio de Ginecología y Obstetricia, Hospital Obrero No2 CNS; bJefe del Servicio de Ginecología y Obstetricia del Hospital Obrero No 2 - CNS;cResidente de tercer año de Ginecología y Obstetricia, Hospital Obrero No2 - CNS.*Correspodencia a:Maita Q. Freddy. Correo electrónico: freddymaita@hotmail.com
Recibido el 20 de junio de 2017.
Aceptado el 12 de septiembre de 2017.
]]>
Resumen
Meckel Gruber es un síndrome letal poco común, descrito por primera vez en 1822 por Meckel; se caracteriza por múltiples malformaciones congénitas que afectan principalmente la cabeza, riñones, dedos de manos y pies. De herencia autosómica recesiva, es reportado comúnmente en matrimonios consanguíneos, también ocurre en las parejas no consanguíneas. Ha sido descrito en todo el mundo, y su incidencia oscila entre 1 por 13 250 – 140 000 nacidos vivos. Comúnmente se diagnostica desde la semana 18 mediante ecografía, aunque se puede hacerlo más tempranamente en familias de alto riesgo. Se reporta el caso clínico de una paciente atendida en el Hospital Obrero No 2 de la Caja Nacional de Salud, sin antecedente de consanguinidad con su pareja, tuvo tres embarazos, de los cuales dos tenían el síndrome de Meckel-Gruber.
Palabras claves:Síndrome de Meckel-Gruber, diagnóstico prenatal, anomalía letal.
Abstract
Meckel-Gruber is an uncommon lethal syndrome, it was described by Meckel in 1822 characterized by multiple congenital malformations, that mainly affects the head, kidneys and fingers; is a autosomal recessive inheritance, reported in consanguineous marriages, but also in non-consanguineous marriages. The worldwide incidence varies from in 13 250 to 140 000 live births. It is usually diagnosed from 18 weeks of pregnancy by ultrasound, although it can be done earlier in high-risk families.
We reported the clinical case of a patient treated in Hospital Obrero No 2 de la Caja Nacional de Salud without a history of consanguinity with her partner with whom she had three pregnancies, of wich two had Meckel-Gruber syndrome.
]]>
Keywords:Meckel-Gruber síndrome, prenatal diagnosis, lethal malformation.
En 1822 Johann Friedrich Meckel describe por primera vez este síndrome, con el hallazgo de dos gemelos quienes murieron con idénticas malformaciones: encefalocele occipital, polidactilia, y riñones poliquísticos1.
En 1934 George B Gruber describió casos familiares de fetos con mismas características clínicas, incluyendo onfalocele, ambigüedad genital y fibrosis hepática; él fue que propuso el término de disencefalia esplacnoquistica a tales hallazgos.
Fueron Opitz y Howe que propusieron el nombre de síndrome de Meckel-Gruber1,3.
El síndrome de Meckel Gruber es un síndrome letal, de característica autosómica recesiva; el riesgo de recurrencia es del 25% en cada embarazo. Ha sido reportado comúnmente en matrimonios consanguíneos pero también ocurre en las parejas no consanguíneas. Tiene penetrancia de 100% y expresividad variable. Una gran variedad de malformaciones han sido observadas en este síndrome siendo los más constantes el encefalocele occipital, los riñones poliquísticos y la polidactilia1-3.
Muchos genes de diferente localización están implicados en la ocurrencia del síndrome. En el síndrome de Meckel Gruber tipo 1 está implicado la mutación de un gen, localizado en el cromosoma 17q22 (MKS1), que codifica componentes proteicos del aparato flagelar y ciliar que causan su disfunción primaria durante la embriogénesis temprana (ciliopatías). También se ha reportado la mutación de la proteína transmembrana Meckelin en el cromosoma 8q (MKS3)4.
Otros loci mapeados que contienen genes mutados están localizados en el cromosoma 11q3 (MKS2); cromosoma 12q (MKS4)5; cromosoma 16q12, 2 (MKS5); cromosoma 4p15 (MKS6); cromosoma 3q22 (MKS7); cromosoma 12q24.31 (MKS8)6,7, otras mutaciones menos frecuentes asociadas. Son estos hallazgos que sugieren la heterogeneidad genética en el síndrome de Meckel Gruber1.
La incidencia mundial oscila entre 1 por 13 250 – 140 000 nacidos vivos. Una alta incidencia ha sido reportada en la población india Guajarati con 1 nacido en cada 3 500 (tasa de portadores de 1 en 18). El síndrome de Meckel Gruber ha sido descrito en todo el mundo8,9. ]]> La tasa de mortalidad en este síndrome es del 100%; el oligoamnios secundario determina una hipoplasia pulmonar que es la principal causa de muerte11. La muerte puede ocurrir intraútero o poco después del nacimiento, esto ha llevado a realizar abortos terapéuticos e interrupciones de embarazos de mayor edad gestacional en los casos diagnosticados4-11.
Puede ser diagnosticado, especialmente en familias con riesgo elevado, ya desde la 11 a 14 semanas mediante la visualización de anomalías en el sistema nervioso central, riñones agrandados e incluso polidactilia; los estudios más tardíos se dificultan por el oligoamnios severo10, aunque en general se la diagnostica en estudios ecográficos de rutina en embarazos de segundo trimestre8.
En todos los casos es necesario realizar el árbol genealógico de la pareja para asesoramiento genético2.
A continuación se reporta el caso de una pareja que presentó dos embarazos con fetos síndrome de Meckel Gruber y un feto normal.
Presentación del caso
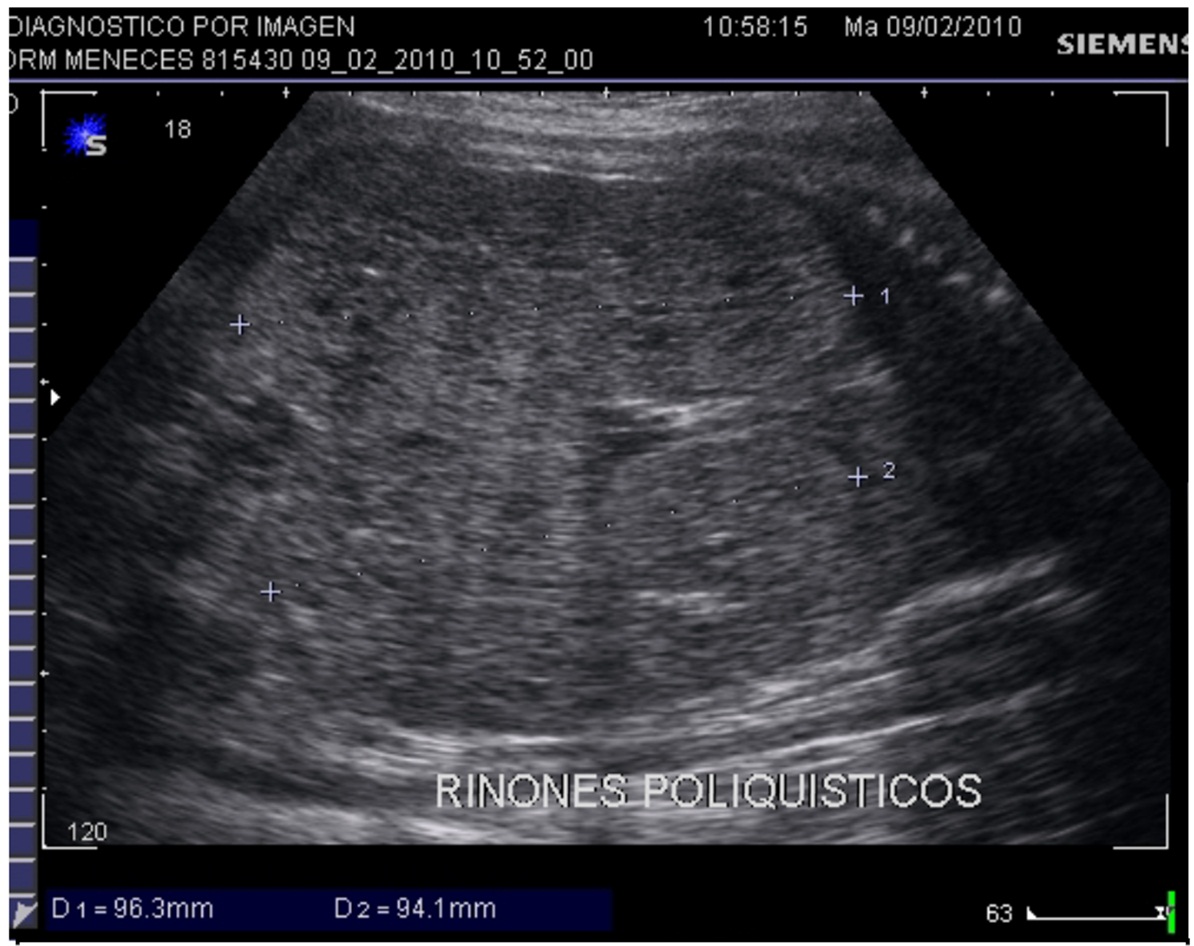
Se describe la historia obstétrica de una paciente que inicia el 2010 a los 30 años con su primer embarazo en el cual, en la semana 34, un estudio ecográfico detectó un feto con polimalformacion que incluía: encefalocele occipital (Figura 1), riñones poliquísticos (Figura 2), pie bot bilateral y oligoamnios severo.
Figura 1:Se muestra el defecto en el hueso occipital por donde se hernia el encéfalo y sus cubiertas.
]]>
Por el cuadro de polimalformativo fetal se determinó terminar el embarazo por cesárea por cuadro compatible con Síndrome de Meckel Gruber; el neonato falleció a los pocos minutos después de nacer.
La apariencia externa confirmó el encefalocele prominente y el pie bot bilateral; no se realizó autopsia por solicitud de los padres. La paciente fue dada de alta al tercer día por buena evolución.
El año 2014 la paciente se embarazo nuevamente de su misma pareja; los estudio ecográficos realizados del embarazo no mostraron alteraciones morfológicas. El embarazó culminó por cesárea por preeclampsia, obteniéndose un producto sano de 3 100 gramos de peso.
A finales del 2015, a los 34 años de edad, la paciente nuevamente se embarazó y fue captada por primera vez cuando acudió al servicio de emergencia de Ginecología y Obstetricia por cuadro gastrointestinal agudo y embarazo.
La anamnesis determinó que tenía 21,1 semanas por clínica y cuadro gastroenteritis aguda; el estudio ecográfico del embarazo mostró un embarazo de 21 semanas, oligoamnios grave, encefalocele occipital (Figura 3), y riñones poliquísticos.
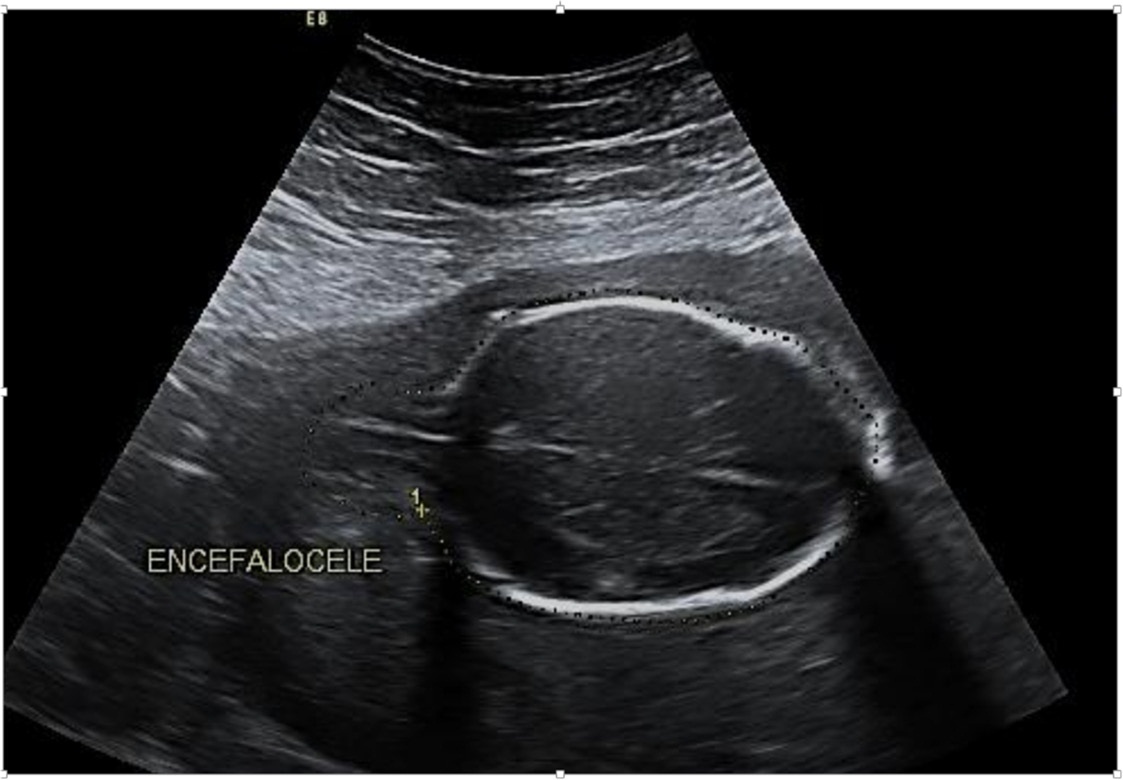
Figura 3:Imagen ecográfica del encefalocele occipital a través del cráneo bífido.
El estudio posterior, ya en el consultorio de medicina fetal, corroboró tales hallazgos y concluyó que se trataba del Síndrome de Meckel-Gruber.
]]> Una vez resuelto el cuadro intestinal de la madre, al tratarse de un embarazo con una patología fetal letal, se realizó el asesoramiento genético correspondiente a la pareja en la Unidad de Medicina Fetal, en esta, no se constató consanguinidad con la pareja, padre de los tres productos. Una junta médica determinó la interrupción del embarazo previo consentimiento informado.La pareja decidió, además, realizar salpingoclasia bilateral como método de planificación familiar definitivo por el riesgo de recurrencia. El embarazo terminó por cesárea, obteniéndose un producto de sexo masculino de 500 gramos de peso que murió al nacer.
El examen clínico posnatal confirmó el encefalocele occipital (Figura 4) y el abdomen globuloso por los riñones poliquísticos, sin embargo, tampoco en esta ocasión los padres dieron su consentimiento para la autopsia.
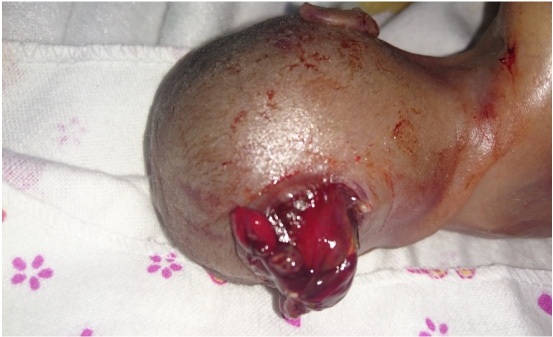
Figura 4:Encefalocele occipital a través del cráneo bífido, evidenciado al nacer.
Discusión
El síndrome de Meckel Gruber es una enfermedad genética, de transmisión autosómica recesiva que tiene una probabilidad de ocurrencia del 25% en cada embarazo en padres portadores. La pareja del caso clínico, tuvo dos hijos afectados de tres, que ha confluido con la rareza de su presentación en padres no consanguíneos.
El diagnóstico prenatal, en nuestro medio, es fundamentalmente ecográfico, en este caso ocurrió después de las 20 semanas que inició con hallazgos anormales en estudios estándar de rutina que llevaron a un estudio más detallado para el diagnóstico, si bien esta descrito que ya en las 11-12 semanas podría ser diagnosticado, especialmente en familias con riesgo elevado mediante la visualización de anomalías en el sistema nervioso central, riñones e incluso en los dedos.
Los criterios diagnósticos de síndrome de Meckel prenatal se fundamentaron al encontrar la presencia de dos de las tres malformaciones clásicas: meningo-encefalocele occipital, displasia renal multiquística y polidactilia postaxial bilateral que se describen que están presentes en 100%, 90% y 83,3% de los casos respectivamente8; otra malformación constante es la fibrosis hepática que no se pudo demostrar. Estos hallazgos son así considerados como criterios mayores para el diagnóstico1-9.
]]> Las malformaciones a nivel del sistema nervioso central son variables; están descritos defectos del cuerpo calloso y de la línea media en general, hidrocefalia, etc. La más frecuente es la microcefalia en los casos con meningoencefalocele occipital. También son frecuentes las disgenesias de la fosa posterior (quistes en fosa posterior, agenesia de vermix, etc.).En todos los casos se recomienda el examen anatomopatológico postnatal para llegar al diagnóstico, esto no fue posible en los dos casos por decisión de los padres. Existen otras múltiples malformaciones descritos que se deben buscar en este síndrome, si bien su frecuencia es menor, por ejemplo, fisura palatina (45% de casos), malformaciones cardiacas (20%), de la lengua, a nivel esplénico, oculares (microftalmia,) displasia retiniana, catarata congénita, etc.), faciales, micrognatia, orejas de implantación baja, boca ancha y labios engrosados1-5.
En los casos de familias con mutación conocida, el diagnóstico genético prenatal es necesario cuando se tiene disponibilidad de estos estudios1-10.
El diagnóstico diferencial debe hacerse con cromosomopatías como la trisomía 13, la trisomía 18, en ambos, la realización de un cariotipo sería diagnostica; con el síndrome de Smith-Lemli-Opitz, el síndrome de Bardet-Biedl, el síndrome de Beemer-Langer, el síndrome de Carpenter-Hunter, el síndrome de Joubert de transmisión autosomica recesiva pero genéticamente heterogéneo, que se asocia con frecuencia displasia renal y polidactilia y muchos otros síndromes polimalformativos.
Los hallazgos que orienten al diagnóstico serán el encefalocele occipital, riñones poliquísticos, la polidactilia y otros hallazgos que serán confirmados por la autopsia.
Conclusión
El síndrome es una enfermedad rara de herencia autosómica recesiva, de mayor frecuencia en parejas consanguíneas, aunque también ocurre en matrimonios no consanguíneos; el diagnostico, en nuestro medio, está basado principalmente en hallazgos ecográficos de segundo trimestre aunque esta descrito que puede realizarse desde la semana 11 en pacientes con riesgo conocido y a través del diagnóstico genético.
El asesoramiento genético es importante ya que existe el riesgo de recurrencia del 25% en cada embarazo, esto permitirá a la pareja decidir sobre su futuro obstétrico.
1. Young ID, Rickett AB, Clarke M. High incidence of Meckel’s síndrome in Gujarati Indians. J Med Genet. 1985; 22:301-4.
]]> 2. Ramadani HM, Nasrat HA. Prenatal diagnosis of recurrent Meckel síndrome. Int J Gynaecol Obstet. 1992; 39: 327-323. Opitz JM, Howe JJ. The Meckel syndrome (dysencephalia splanchnocystica, the Gruber syndrome) Birth Defects. 1969; 2: 167-79
4. Smith UM, Consugar M, Tee LJ, Mckee BM, Maina EN, Welan S. The transmembrabe protein Meckelin (MKS3) is mutated in Meckel Gruber syndrome and the wpk rat. Nat Genet. 2006 Feb. 38(2): 191-6 [medline]
5. Tallila J, Jakkula E, Peltonen L, Salonen R, Kestila L. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliophaty puzzle. Am J Hum Genet. 2008 Jun. 82(6): 1361-7
6. Online Mendelian Inheritance in Man. Meckel syndrome, type 7; MKS7. OMIN
7. Online Mendelian Inheritance in Man. Meckel syndrome, type 8; MKS8. OMIN
8. Parelkar SV, Kapadnis SP, Sanghvi BV, Joshi PB, Mundada D, Oak SN. Meckel Gruber syndrome: A rare and lethal anomaly with review of literatura. J Pediatr Neurosci. 2013 May 8(2): 154-7. [medline]
9. Shetty PB, Alva N, Patil S, Shetty R. Meckel-Gruber Syndrome (dysencephalia splanchnocystica). J Contemp Dent Pract 2012 Sep 1. 13(5): 713-5.
10. Sepulveda W, Sebire NJ, Souka A, Snijders RJ, Nicolaides KH. Diagnosis of Meckel Gruber syndrome at eleven to fourteen weeks’ gestation. Am J Obstet Gynecol. 1997; 176: 316-9.
11. Barisic I, Boban L, Loane M y cols. Sindrome de Meckel-Gruber: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. Eur J Hum Genet. 2015 Jun. 23 (6): 746-52. [medline] ]]>