











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
Permalink
INTRODUCCION
La quinua es un pseudo cereal originario de América del Sur, principalmente de los andes de Bolivia y Perú, es considerado de alto valor nutricional debido al contenido de ácidos grasos, oligoelementos y proteínas (13.81-21.90% dependiendo la variedad) enriquecidas en aminoácidos esenciales superiores a otros cereales como el trigo, cebada y soya (Bojanic, 2011). Además de sus propiedades nutricionales, la quinua ha despertado su interés de estudio por su amplia adaptación a distintas condiciones agroecológicas y una diversidad en cuanto a sus características genéticas que influyen en la expresión de determinadas características morfológicas, fenológicas y principalmente metabólicas, que van en relación al tipo de estrés al cual está sometida la planta (biótico o abiótico) (Jarvis et al., 2017; Morales, Garcia et al., 2013; Mujica & Jacobsen, 2006; Rojas et al., 2016).
Estudios a la fecha (Garcia E. et al., 2018; Méndez Espinoza & Vallejo Reyna, 2019), han demostrado que los mecanismos de respuesta en las plantas a diferentes tipos de estrés abiótico (sequia, salinidad y frio) o bióticos (ataque de plagas, defensa de predadores, etc.), son tres y se clasifican como: mecanismos constitutivos, defensa pasiva a los agentes patógenos; estructurales constitutivos, acumulación de ceras, formación de vellosidades, etc. y químicos constitutivos, acumulación de metabolitos secundarios (Madriz O., 2002; Nakashima et al., 2009). Todas estas modificaciones en el organismo vegetal, facilitan una mejor captación de la señal de estrés (adaptación al medio) y estas llegan a modificar la expresión génica mediante la función reguladora de los factores de transcripción, donde existe la unión de proteínas especificas con elementos cis del ADN, que son fragmentos de nucleótidos no codificantes, contiguos a los genes blancos o de interés (Morales et al., 2013). Los estudios científicos de bio-prospección, quimio-taxonomía, y otros. han sido realizados con éxito y claridad haciendo uso de las técnicas de biología molecular y la bioinformática, que facilitan la identificación de genes involucrados en la expresión del metabolismo y/o regulación de la biosíntesis, a partir de material genético secuenciado o mediante una secuenciación de Novo y haciendo uso de una de sus técnicas más aplicadas denominado PCR (reacción en cadena de polimerasa) (Angarita et al., 2017; Jiménez et al., 2017; Palacios et al., 2004).
El 9 de febrero de 2017, fue hecha pública la noticia en la plataforma de la BBC NEWS, sobre la secuenciación completa del genoma de la quinua por un grupo de investigadores a la cabeza Mark Tester de la Universidad del Rey Abdullah de Ciencia y Tecnología (KAUST) en Arabia Saudita (McGrath, 2017). Así mismo, estimaron que el genoma de la quinua tiene alrededor de 44.776 genes que fueron determinados por la técnica SMRT (Single Molecule Real Time) (Rodriguez, 2017). El mismo año en la publicación de Jarvis et al., 2017, se realizó el trabajo de la secuenciación y ensamblaje del genoma de una variedad de quinua chilena costera, aplicando la técnica de SMRT (figura 1). Donde se determinó que el ensamblaje total del genoma equivale a 1,39 giga-bases (Gb) y a su vez, se determinó que los genes involucrados en la expresión de las proteínas y los micro ARNs predichos corresponden de igual manera al reportado por el grupo de investigación de Mark Tester.
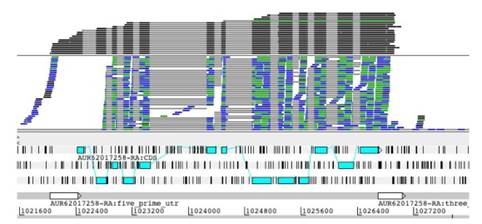
Figura 1 Secuenciación por isoformas (ARN-seq) del genoma de la quinua. Resultado del mapeo para el material genético de la quinua costera chilena, empleando la técnica de secuenciación por isoformas, donde se aprecia el ensamblaje total del material genético en estudio. Fuente: Jarvis et al., 2017.
La gran cantidad de variedades de quinuas entre dulces y amargas, resistentes a los agentes bióticos y abióticos, hacen también que exista una gran diversidad en cuanto al material genético (Serna et al., 2020) razón por la cual se han desarrollado los bancos de germoplasma (Rojas et al., 2013).
Un daño que ocasiona pérdidas en los cultivos, es un agente patógeno denominado Mildiu (Peronospora variabilis) (ver figura 2), una de las formas de control de este patógeno es la aplicación por riego de metalaxyl (fungicida). Sin embargo, existen algunas variedades que adquirieron inmunidad genética (Danielsen & Ames, 2007); y es así que en el trabajo realizado por (Colque-Little et al., 2021), realizó la infección de 132 accesiones de quinua sudamericana con P. variabilis y se hizo un control del ambiente de contagio dentro un invernadero independiente. En el cual se evidenció que los niveles de contagio variaron entre 5 a un 86%, esta estimación permitió diferenciar entre las variedades susceptibles y las variedades con resistencia al patógeno. Por otro lado, el hecho de no haber descubierto aun los genes responsables para la resistencia a este patógeno, esta propiedad puede ser aplicada a otras variedades susceptibles por fito mejoramiento.
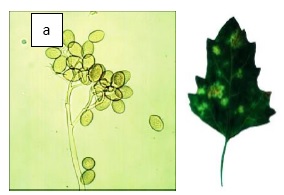
Figura 2 Agente patógeno de la quinua. a) Mildiu (P. variabilis) y b) hoja de quinua infectada. a. Fotografía en un microscopio electrónico del agente patógeno que afecta a la quinua y b) fotografía de la apariencia de una hoja infectada. Fuente: Colque-Little et al., 2021.
En cuanto al contenido de saponinas, varía de acuerdo a la etapa fenológica y en la quinua se han encontrado hasta el 2016 alrededor de 30 saponinas entre mono y bi-desmosidícas con siete tipos de anillos pentacíclicos derivados de la β-amirina (figura 3, Ahumada et al., 2016).
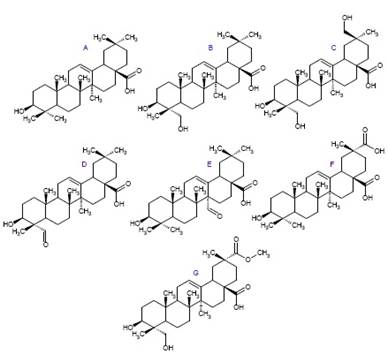
Figura 3 Sapogeninas de quinua encontradas en distintas partes de la planta. A) Ac. Oleanólico, B) Hederagenina, C) Acido 3β, 23, 30-trihidroxi olean-12-eno-28-oico, D) Gipsogenina, E) Ac. 3β-hidroxi-27-oxoolean-12-eno-28-oico, F) Ac. Espergulagenico, F) Ac. Serjánico y G) Ac. Fitolacagénico.
Nuevamente indicamos que el contenido de saponinas en quinua las clasificara entre variedades dulces y amargas. y al ser de naturaleza anti-nutricionales, estas deben ser eliminadas antes de su consumo (Fiallos-Jurado et al., 2016; Serna et al., 2020). En la actualidad, muchos trabajos de investigación aplican las técnicas de biología molecular para la identificación de genes involucrados en la biosíntesis de saponinas.
Es así que en el trabajo de James R. (2009), se realizó la secuenciación de la quinua empleando el método de Sanger y la pirosecuenciación (secuenciación 454). Por un lado, el método Sanger realizó 18.325 lecturas con un promedio de 693 nucleótidos por lectura. Mientras que con el método de pirosecuenciación se obtuvo 298.048 lecturas con un promedio de 202 nucleótidos por lectura, con estos resultados obtenidos se realizó un ensamblaje hibrido entre las dos técnicas aplicadas generando así un conjunto unigene del cual fueron identificado 291 nuevos marcadores de micro satélites al cual se aplicó la técnica de microarray para posteriormente realizar las comparaciones entre variedades dulces y amargas. Como resultado de su investigación, se identificaron genes que están involucrados en la expresión de compuestos triterpenóides; por otro lado, también fueron identificados genes que son homólogos al citocromo P450s, las monooxigenasas del citocromo P450 y las glicosiltransferasas (figura 4). Como recomendación final establece que estos genes identificados sean tomados en cuenta al momento del estudio de la biosíntesis de saponinas
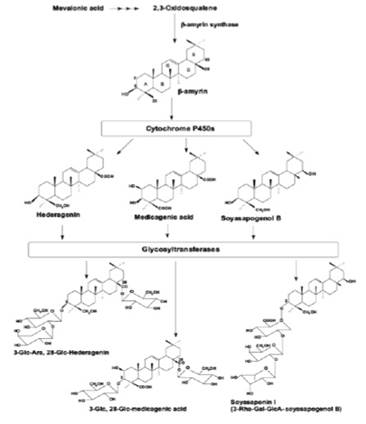
Figura 4 La ruta de biosíntesis de saponinas en quinua. Representación esquemática de la ruta de biosíntesis de saponinas de quinua bajo la influencia de genes homólogos del citocromo P450s y enzimas glucosil transferasas. Fuente: James R., 2009.
Por otro lado, en la investigación realizada por Fiallos-Jurado et al., 2016, se evidenció que el contenido de saponinas en hojas de dos variedades de quinua (dulce y amarga) se encontraba regulada a través de una fitohormona, denominada MeJa (Metil jasmonato). El estudio consistió en el desarrollo de ambas variedades de quinua inmersas a distintos tiempos (2, 24 y 48 h) con la fitohormona y al momento de la cuantificación de saponinas se observó que existía una variación en la producción de este metabolito (figura 5).
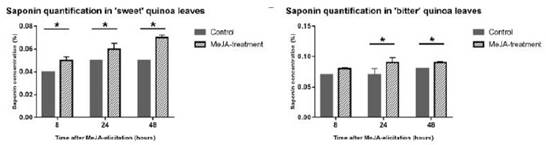
Figura 5 Cuantificación de saponinas en hojas de quinua tratadas con MeJa (jasmonato de metilo). Representación gráfica de la expresión de saponinas en quinuas dulces y amargas por el tratamiento con MeJa. En ambos casos se observa un incremento en el contenido de saponinas con el paso del tratamiento, sin embargo en la quinua dulce se aprecia un mayor incremento a las 48 horas de exposición mientras que la quinua amarga no tiene mayor cambio en su contenido de saponinas a las 24 y 48 horas respecto a los controles. Fuente: Fiallos-Jurado et al., 2016.
Es en relación a la figura 5 la variedad de quinua dulce, tuvo una pronta respuesta al tratamiento con MeJa (8 horas). Sin embargo, la quinua amarga mostro una respuesta positiva pasadas las 24 horas. Así, con base en este análisis preliminar los autores tenían la idea que la biosíntesis de saponinas se encontraba regulada por la enzima bAS que es el marcador para la producción de ácido jasmónico en presencia de un agente estresor (Laredo Alcalá et al., 2017; Tijet & Brash, 2002).
Posteriormente se realizó un análisis a nivel del transcriptoma para la identificación de genes que eran activados por MeJa, de manera inicial se vio el gen de la β-amirina sintasa caracterizada de Maesa lanceolata, determinando que para la quinua esta enzima es ortóloga; es decir, que la quinua presenta homología con otras especies vegetales (Contreras, 2015). La enzima bAS (figura 6), denominada como el gen CqbAS1, también fue evaluada dentro de las variedades de quinua mediante PCR-q, obteniendo los mismos resultados que en la cuantificación de saponinas. Pues la variedad de quinua dulce tiene una respuesta más rápida que la quinua amarga.
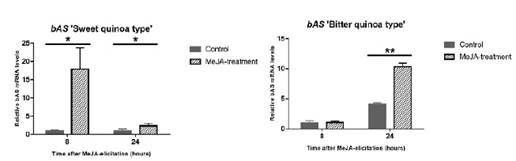
Figura 6 Aumento de los niveles de la enzima bAS en hojas tratadas con MeJa. Representación gráfica de los niveles de expresión de la enzima bAS bajo tratamiento con MeJa. En ambos casos se tiene variaciones durante el tratamiento, como resultado se aprecia que la expresión de la enzima bAS es más rápida en la quinua dulce tras las 8 horas de exposición, sin embargo esta disminuye drásticamente pasadas las 24 horas pero incrementa su contenido en la quinua amarga respecto a los controles. Fuente: Fiallos-Jurado et al., 2016.
Además para una mejor apreciación de las características del gen, este fue expresado mediante clonación, donde se vio que dicho gen es una proteína de 763 aminoácidos que se encarga del proceso de ciclación del escualeno a derivados de la β-amirina. Posteriormente, para las modificaciones que se realizan en las geninas por la adición de mono u oligosacáridos se analizaron las enzimas del citocromo P450 a través de la codificación de la familia de proteínas CYP716A12 de M. truncatula dentro en el transcriptoma de la quinua, aplicando la plataforma TBLASTX se logró identificar 3 genes candidatos de la P450: CYP716A78, CYP716A79 y CYP716AB1. Estos al ser analizados por PCR-q (figura 7) se reportó el mismo comportamiento de los anteriores casos.
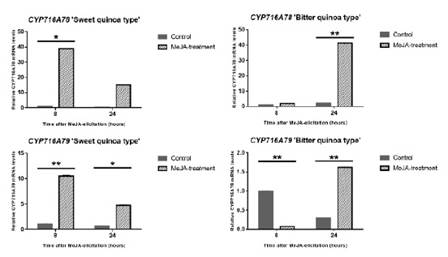
Figura 7 Aumento de los niveles de los genes candidatos en la expresión de saponinas en hojas tratadas con MeJa. Representación gráfica de los genes candidatos responsables a la produccion de saponinas de quinua bajo tratamiento con MeJa. Ambos genes CYP716A78 y CYP716A79 en variedades de quinua dulce y amarga presentan variaciones, pero como ya se vio en las anteriores graficas la quinua dulce tiene pronta respuesta (8 horas) al tratamiento con MeJa pero disminuye con el pasar del tiempo (24 horas), mientras que la quinua amarga tiene una respuesta tardía produciendo mayores cantidades a las 24 horas del tratamiento. Fuente: Fiallos-Jurado et al., 2016.
METODOLOGIA
La metodología empleada para el presente articulo de revisión bibliográfica fue mediante el uso de: tesis de maestría desarrollado por Derrick James Reynolds (2009) (Department of Plant ans Wildlife Sciences-Brigham Young University), artículos científicos (plataformas: Scielo, Redalyc, Elsevier, Springer, entre otras), libros y plataformas WEB (www.bbc.com, www.fao.org, www.sidalc.net y https://biología.laguía2000.com) con principal interés en: genoma de la quinua, técnicas de secuenciación, genes identificados y su posterior expresión génica.
PCR (Chain Reaction Polymerase)
Esta técnica es ampliamente utilizada para la amplificación de fragmentos o secuencias cortas de ADN (cebadores) que son de interés de estudio, en las revisiones realizadas la técnica es importancia para la amplificación de los genes de interés (CYP716A78 y CYP716A79) identificados como responsables de la síntesis de saponinas en variedades de quinua dulce y amarga como lo realizó Fiallos-Jurado et al., 2016. El principio de la técnica se basa principalmente en la formación de secuencias complementarias al fragmento de ADN o molde que mediante una variación de temperatura las desnaturalizan y separan, este proceso ayuda a las enzimas involucradas con este proceso de replicación haciendo que en poco tiempo pueda generarse muchas copias (Rådström et al., 2004; Smith & Osborn, 2009).
SMRT (Single Molecule Real Time)
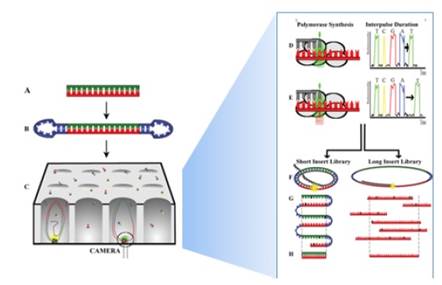
Esta técnica de secuenciación ha sido desarrollada principalmente por: Pacific Biociences (PacBio) y además mediante la tecnología de nanoporos de Oxford Nanopore Technologies. La técnica tiene un interés de aplicación cuando se habla de una secuenciación de Novo, ya que esta técnica permite la secuenciación de material genético extenso con un nivel de confianza del 99.999% (Ardui et al., 2018), sin embargo también se ha reportado algunos errores que afectan a su sensibilidad pero este puede ser minimizado con la adaptación de otros métodos de secuenciación (Roberts et al., 2013). Su aplicación consiste básicamente en 8 pasos en los que se realiza la secuenciación del ADN (Ardui et al., 2018). Es así que la técnica fue tomada como una opción para la secuenciación del genoma de la quinua, pues aún no se conocía el tamaño de su material genético y como resultado final se determinó que está compuesto por 44.776 genes (Rodríguez, 2017).
Figura 8: Técnica de secuenciación por SMTR desarrollado por PacBio y Oxford Nanopore Technologies. Esquema de la secuenciación SMTR aplicado al estudio genómico de la quinua desarrollado en la investigación de Rodríguez, 2017. Fuente: Ardui et al., 2018
Secuenciación RNA-seq
La técnica de RNA-seq o secuenciación de isoformas, nos indica los niveles de desarrollo, tratamientos y condiciones de distintos tejidos, órganos u organismos desde su estado del transcriptoma. es por tal motivo que los autores incluyen esta técnica de secuenciación a través del RNA pues a nivel del post transcritoma se puede llegar a determinar la secuencia del material genético como lo hizo Jarvis et al., 2017 en su publicación determinado que el material genético se encuentra compuesto por 1,39 giga bases. Así pues, esta técnica es un método de secuenciación cuantitativa que inicia desde la extracción de ARN-total que es una mezcla de ARN-m y ARN-r que se encuentra en mayor proporción. De esta manera, el análisis se realiza a nivel del ARN-m, donde debe ser purificado mediante técnicas establecidas, una de ellas es la purificación de ARN-m por la captura positiva con ARN poliadenilado (PolyA capture) y la segunda técnica es la eliminación del ARN-r (ARN depletion). La aplicación de estas técnicas dependerá si se encuentra trabajando con organismos eucariontes o procariontes (Rodriguez A. & Shishkova, 2018).
Figura 9. Principio de secuenciación por ARN-seq. Esquema de secuenciación por isoformas o ARN-seq para la identificación del genoma de la quinua aplicado por Jarvis et al, 2017 en su investigación. Fuente: Rodriguez A. & Shishkova, 2018.
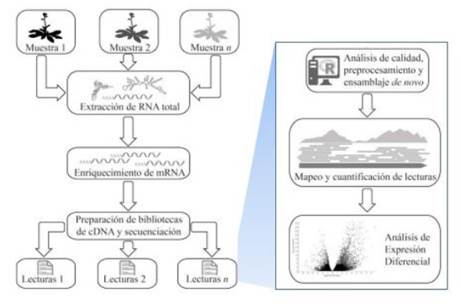
Posteriormente el ARN-m se aplica para la creación de bibliotecas con el que se podrá dar inicio a la secuenciación del ADN completo o secuenciarlo por fragmentación (fragmentos de 200 a 1000 nucleótidos), este último es el más común por su bajo costo. Cada fragmento que ha sido secuenciado (reads) pasa a ser parte del mapeo y estos pasan a cuantificar la abundancia de cada transcrito (Rodriguez A. & Shishkova, 2018).
Secuenciación de Sanger
Esta técnica fue desarrollada por Frederik Sanger en 1977 y desde entonces muchas de las técnicas desarrolladas hasta la actualidad son modificaciones a su propuesta (Sanger et al., 1977). El principio de la secuenciación de esta técnica consiste en siete pasos (Gauthier, 2007):
Figura 10. Fundamento de la secuenciación de Sanger. Esquema de la secuenciación de Sanger, aplicado en la investigación desarrollada por James R., 2009 para la obtención hibrida (técnica de secuenciación combinada con la técnica de pirosecuenciación) del material genético de la quinua donde obtuvo 693 nucleótidos. Fuente: Gauthier, 2007.
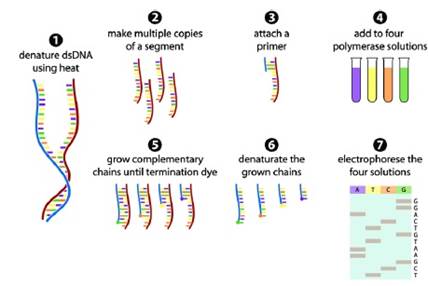
Sin embargo al ser esta una técnica desarrollada hace muchos años atrás, presenta algunas desventajas respectos a nuevos métodos desarrollados (Fakruddin & Abhiiit, 2012). Es por tal motivo que durante la investigación el autor hace referencia a la combinación de la secuenciación de Sanger con la pirosecuenciación.
Pirosecuenciación
Es una novedosa técnica de secuenciación del ADN que ha sido desarrollada por Royal Institute Technology KTH y es un método alternativo al método de Sanger para la secuenciación de Novo por las ventajas que esta técnica ofrece, entre ellas tenemos: precisión, flexibilidad, fácil automatización y en muchos casos se han utilizado como técnica de confirmación para una secuenciación de Novo, es así que James R., 2009 aplica esta técnica con este fin, combinándola con la secuenciación de Sanger y obtener así una secuencia del material genético de la quinua completa e identificada. Si bien es una técnica lumínica, esta técnica no tiene la necesidad de usar marcadores fluorescentes en cebadores o nucleótidos (Fakruddin & Abhiiit, 2012).
Esta técnica se basa en la secuenciación por síntesis (ver figura 11), partiendo de un ADN molde (monocatenario marcado con biotina) al cual se une un cebador y para que este de inicio a la síntesis debe unirse a cuatro enzimas: ADN polimerasa, ATP sulfurilasa, luciferasa y apirasa. Además de estas enzimas, se debe adicionar desoxinucleotidos trifosfatos (dNTP), entonces se da inicio a la síntesis y con cada adición de nucleótido por la polimerasa se produce la liberación de una molécula de pirofosfato (PPi) inorgánico.
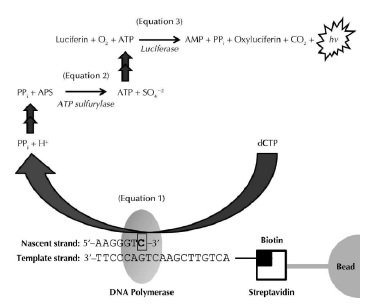
Figura 11 Esquema de la técnica de pirosecuenciación en base a la síntesis de Novo material genético. Esquema de la pirosecuenciación aplicada para la obtención de una secuencia hibrida del material genético de la quinua desarrollada por (James R., 2009) en su investigación donde obtuvo como promedio 202 nucleótidos por lectura realizada (298.048) Fuente: Harrington et al., 2013.
Una ves que el PPi es liberado, este se convierte cuantitativamente en ATP mediante la ATP-sulfurilasa en presencia de APS (Adenosina Fosfosulfato-sustrato), la cantidad de ATP generada es catalizada por la luciferasa produciendo una la luz visible (560nm)(Iliyina et al., 1998) que es detectado por un fotomultiplicador o un CCD. Finalmente la luz generada y posteriormente detectada, genera una señal (pico) en el pirograma que es proporcional a la cantidad de nucleotidos incorporados en la sintesis (Fakruddin & Abhiiit, 2012; Harrington et al., 2013).
CONCLUSIONES
La quinua al ser un alimento importante debido a su contenido de ácidos grasos, oligoelementos y proteínas con alto contenido en aminoácidos esenciales (Bojanic, 2011), además de su contenido de metabolitos secundarios como las saponinas que son producidas como mecanismo de defensa ante los agentes patógenos (Ahumada et al., 2016; Bruneton, 1993). Sin embargo, este tipo de metabolitos son anti nutricionales para el organismo por lo que deben ser eliminados por un proceso adicional: el escarificado (Ahumada et al., 2016; Candia D., 2016).
En la actualidad aún se desea emplear estas técnicas como alternativas genéticas para la eliminación de saponinas y evitar el paso adicional del escarificado (Fiallos-Jurado et al., 2016). Así mismo, el año 2017 con el ensamblaje completo del genoma de la quinua por el método SMTR (Jarvis et al., 2017), ha sido el punto de partida para el descubrimiento de genes involucrados en la biosíntesis de saponinas aplicando diferentes técnicas de biología molecular (ARN-seq, PCR-q, etc.) y el uso de plataformas como el TBLASTX.
En dos ocasiones se han establecido posibles rutas para la biosíntesis de saponinas en la quinua. El 2009, en el estudio de una tesis doctoral se ha concluido que la producción de saponinas están involucradas las enzimas análogas del citocromo P450 y enzimas de tipo glucosil transferasas (James R., 2009). y posteriormente en un estudio realizado el año 2016, se llegó a la misma conclusión; sin embargo, este sería expresado por los genes CYP716A78, CYP716A79 y CYP716AB1 (perteneciente al grupo del citocromo P450), además que su nivel de expresión varia en relación a la producción de enzimas marcadoras del ácido jasmonico (Fiallos-Jurado et al., 2016)
Por otro lado, los niveles de expresión génica ante los factores bióticos como el mildiu, aún no han sido reportados pero en el estudio realizado por Danielsen & Ames, 2007 indica que esta propiedad (resistencia al mildiu) puede ser transmitida mediante técnicas de Fito mejoramiento convencional y además que los niveles en la producción de saponina no se encontrarían involucrados en la resistencia a este patógeno

