










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO  uBio
uBio Compartir

 Permalink
PermalinkRevista CON-CIENCIA
versión impresa ISSN 2310-0265
Rev.Cs.Farm. y Bioq v.8 n.2 La Paz nov. 2020
ARTÍCULOS ORIGINALES
Estudio de Bioequivalencia in vitro de comprimidos de liberación inmediata de
Metformina de 850 mg comercializados en Bolivia
In vitro Bioequivalence study of immediate release tablets of 850 mg
of Metformin marketed in Bolivia
Giovana Ximena Saravia Gutierrez1![]() , María Luisa Daza Calderón2
, María Luisa Daza Calderón2![]()
1 Facultad de Ciencias Farmacéuticas y Bioquímicas, Universidad Mayor de San Andrés
La Paz-Bolivia, gxsg@hotmail.com
2 Facultad de Ciencias Farmacéuticas y Bioquímicas, Universidad Mayor de San Andrés
La Paz-Bolivia, mldazacal@hotmail.com
Fecha de recepción: 29 Septiembre 2020 Fecha de aceptación: 18 Octibre de 2020
Resumen
Introducción: Los estudios de Bioequivalencia demuestran la equivalencia en la calidad biofarmacéutica entre el producto farmacéutico multifuente y el medicamento de referencia, permitiendo el establecimiento de puentes entre las pruebas preclínicas y los ensayos clínicos asociados con el medicamento de referencia. La Metformina clorhidrato es el fármaco de primera elección para el tratamiento de la diabetes mellitus II y pertenece a la clase 3 del Sistema de Clasificación Biofarmacéutica.
Objetivo: Establecer mediante pruebas de disolución in vitro, si 4 productos farmacéuticos multifuentes de liberación inmediata, de administración peroral, con Metformina clorhidrato, comercializados en Bolivia son bioequivalentes en relación con el producto de referencia, a través de un diseño experimental, con significancia estadística.
Método: Se elaboraron perfiles de disolución de cada producto, tomando en cuenta las condiciones del método del ensayo de disolución establecido en la monografía Metformina, tabletas de la USP 39, considerando seis tiempos de muestreo. Se realizó el análisis cinético de los datos de porcentaje disuelto acumulado de los perfiles de disolución versus el tiempo. Se empleó el método de modelo independiente para comparar los perfiles de disolución de los productos a través del cálculo de los factores de diferencia (f1) y de similitud (f2) recomendados para estudios de bioexención por la Organización Mundial de la Salud.
Resultados: Todos los productos tomados en cuenta en la investigación cumplieron satisfactoriamente los análisis de Control de Calidad. Los resultados del estudio de Bioequivalencia mostraron que existen diferencias estadísticamente significativas entre los productos del estudio.
Conclusión: En este estudio no se demostró la Bioequivalencia in vitro de los productos B, C, D y E con relación al producto de referencia A, con base en pruebas de disolución in vitro.
Palabras clave: Bioequivalencia in vitro, Metformina, Multifuente, bioexención.
Abstract
Introduction: Bioequivalence studies demonstrate the equivalence in biopharmaceutical quality between the multi-source pharmaceutical product and the reference medicine, allowing the establishment of bridges between preclinical tests and clinical trials associated with the reference medicine. Metformin hydrochloride is the drug of first choice for the diabetes mellitus II treatment and belongs to class 3 of the Biopharmaceutical Classification System.
Objective: Establish by in vitro dissolution tests, whether 4 multisource pharmaceutical products of immediate release and peroral administration, with Metformin hydrochloride, marketed in Bolivia are bioequivalent in relation to the reference product, through an experimental design, with statistical significance.
Method:
Dissolution profiles were prepared for each product, taking into account the conditions of the dissolution test method established in the "Metformin, tablets" monograph of USP 39, considering six sampling times. The kinetic analysis of the accumulated dissolved percentage data of the dissolution profiles versus time was performed. The independent model method was used to compare the dissolution profiles of the products by calculating the difference factor (f1) and similarity factor (f2) recommended for Biowaiver studies, by the World Health Organization.
Results: All the products taken into account in the investigation, satisfactorily fulfilled the Quality Control analysis. The results of the Bioequivalence study showed that there are statistically significant differences between the products of the study.
Conclusion: In this study the in vitro bioequivalence of the products B, C, D and E in relation to the reference product A was not demonstrated, based on in vitro dissolution tests.
Keywords: In vitro Bioequivalence, Metformin, Multisource, Biowaiver.
INTRODUCCIÓN
La detección empírica de fallas en la terapéutica de ciertos medicamentos ha motivado la implementación de normas para mejorar la calidad de los productos farmacéuticos (Ponce D`León y Jaramillo, 2004, p. 70).
El costo de la atención médica ha aumentado a nivel mundial durante las últimas dos décadas, y esto ha provocado esfuerzos en la mayoría de los países para reducir esos costos. Se sabe que la mayoría de las intervenciones de atención médica se realizan a través de medicamentos. Dado que el costo de la medicación también se ha incrementado a través de los años, la contribución de los costos de los medicamentos a los costos generales de la atención médica ha recibido una atención considerable. Una estrategia importante para reducir el costo de la medicación y, por lo tanto, reducir su contribución al costo total de la atención médica en los mercados mundiales, ha sido la introducción de productos farmacéuticos multifuentes (PFMF) equivalentes a los medicamentos innovadores. La estrategia ha sido efectiva. El ahorro promedio nacional a través del uso de PFMF desde 1997-2000 fue de aproximadamente 9 mil millones de dólares o el 11% de los costos totales de prescripción. Al mismo tiempo, los PFMF han capturado más del 65% del mercado farmacéutico mundial. (Midha y McKay, 2009)
La disponibilidad de diferentes formulaciones de la misma sustancia farmacológica administrada con la misma concentración y en la misma forma de dosificación plantea un desafío especial para los profesionales de la salud, lo que hace que estos temas sean muy relevantes para los farmacéuticos en todos los entornos de práctica (Makoid, Vuchetich y Banakar, 1999, p. 8-2).
El principal papel y la responsabilidad de los farmacéuticos implican el juicio sobre la calidad del medicamento a través de la selección del producto entre las marcas comerciales de fármacos disponibles. Este incluye informes de selección de productos disponibles de distintos fabricantes y sustitución de un producto por otro, cuando se produce un cambio de original al PFMF, de PFMF a original o de PFMF a otro PFMF. Aún la uniformidad de lote a lote en un fabricante puede influir en las consideraciones de calidad de un producto. Un factor significativo en estas decisiones podría ser el potencial ahorro de costos para el paciente (Malinowski, 2003, p. 1155).
La calidad de los medicamentos sean estos PFMF o no, debe ser garantizada por estudios de Bioequivalencia (BE) con el comparador o innovador para poder asegurar su intercambio, en armonía con las directrices de la Organización Panamericana de Salud (OPS) y la Organización mundial de Salud (OMS), favoreciendo su acceso y fortaleciendo las Políticas de PFMF. (Placencia, 2010, p.4)
Para garantizar la intercambiabilidad, el PFMF debe ser terapéuticamente equivalente al producto de comparación (WHO, 2005a, p. 4). Para demostrar la BE de un medicamento se requiere la realización de estudios in vivo en voluntarios sanos (Baena y Ponce D´León, 2008, pp. 18). La demostración práctica directa de la equivalencia terapéutica en un estudio clínico generalmente requiere un gran número de pacientes. Tales estudios en humanos pueden ser financieramente desalentadores, muchas veces innecesarios y en ciertos casos pueden no ser éticos (WHO, 2005a, p. 4).
Es por ello que, se acude a la bioexención, basada en el Sistema de Clasificación Bioarmacéutica (SCB), como una alternativa para demostrar la equivalencia terapéutica, empleando estudios comparativos de liberación-disolución in vitro, basados en la generación de perfiles cinéticos de disolución (Instituto de Salud Pública de Chile, 2007, pp. 16, 27).
En Bolivia, el Artículo 1 de Ley del Medicamento Nª 1737 señala que se debe: Disponer de medicamentos que garanticen inocuidad, eficacia y calidad demostrada, evitando la presencia de fármacos de dudosa calidad, ineficiencia farmacológica o de riesgo terapéutico (Honorable Congreso Nacional de Bolivia, 1996, p.1). Se considera como objetivo esencial, demostrar la eficacia y calidad de los PFMF reconocidos por dicha ley, asegurando su BE con el producto de referencia, demostrando así su equivalencia terapéutica, para proporcionar esta información tanto al prescriptor como al farmacéutico para que pueda intercambiar los productos de referencia con estos PFMF.
La diabetes es una enfermedad crónica, silenciosa, que si no se trata a tiempo puede ser mortal; se caracteriza por presentar niveles aumentados de azúcar en sangre (glucemia). Una enfermedad crónica es aquella que no se cura, pero con seguimiento y tratamiento adecuado la persona con diabetes puede prevenir complicaciones y llevar una vida normal (Ministerio de Salud de la República de Argentina, s.f.).
En la última década se ha registrado que la diabetes mellitus tipo 2 no sólo es ya un problema de adultos, también se está viendo reflejada en adolescentes, lo que complica aún más el problema, convirtiendo esta patología en un serio padecimiento mundial de grandes proporciones. En los últimos diez años, la prevalencia de la diabetes aumentó rápidamente en los países de bajos ingresos. Según proyecciones de la Organización Mundial de la Salud (OMS), la diabetes será la séptima causa de muerte en 2030 ( ). En Bolivia los casos de Diabetes Mellitus se incrementaron de 98100 en 2015 a 138124 en 2016 ( ). Asimismo, el Sistema Nacional de Información en Salud (SNIS) estima que en Bolivia la prevalencia de diabetes es de 6,6 % lo que quiere decir que 362000 personas vivirían con la enfermedad, lo que significaría que cada año mueren cerca de 5260 personas entre 20 y 79 años por causa de la diabetes ( ) Al ser la diabetes una importante causa de ceguera, insuficiencia renal, infarto de miocardio, accidente cerebro vascular y amputación de los miembros inferiores, se hace imprescindible la prevención diagnóstica y el tratamiento oportuno (Ministerio de Salud del Estado Plurinacional de Bolivia, 26 de septiembre de 2017).
Nuestro papel como químicos farmacéuticos especialistas en el medicamento, nos impone el reto de realizar este estudio, para fortalecer nuestras competencias en la dispensación y la capacidad de intercambiar un medicamento de referencia por un PFMF.
El presente trabajo pretende establecer si existe BE in vitro entre 4 PFMF de liberación inmediata y de administración peroral, con metformina clorhidrato como principio activo, comercializados en el mercado boliviano y su correspondiente producto de referencia; con el propósito de comprobar que serán administrados de manera segura y efectiva, al igual que cuando se administra el medicamento de referencia, asegurando el efecto terapéutico normoglucemiante deseado en el tratamiento de la Diabetes Mellitus 2 (DM2).
Se escogieron al azar medicamentos con metformina clorhidrato como principio activo, por ser el medicamento de atención primaria en el tratamiento de la DM2 que es una enfermedad crónica, y porque está incluido en la Lista Nacional de Medicamentos Esenciales LINAME (Ministerio de Salud del Estado Plurinacional de Bolivia,2016).
La metformina clorhidrato pertenece a la clase 3 (de alta solubilidad y baja permeabilidad) del Sistema de Clasificación Biofarmacéutica (SCB), por lo que, según la OMS y la ICH, aplicando pruebas comparativas in vitro de disolución/liberación, se puede asegurar la BE y por consiguiente la equivalencia terapéutica de los productos en estudio: 4 PFMF de liberación inmediata, de administración peroral, con 850 mg de Metformina clorhidrato y el producto de referencia Glucophage®, que son comercializados en Bolivia, corroborando que el valor hallado del factor de similitud (f2) sea mayor a 50.
METODOLOGÍA
Materiales: Metformina HCl (estándar), Fosfato de monobásico de potasio anhidro (Biopack), Hidróxido de sodio p.a. (Merck), Ácido fosfórico al 86,2% (JT Baker Chemical Co.), Agua destilada y agua ultrapura (obtenida a través de un sistema Milli-Q de Millipore).
Equipos: Balanza analítica GR-200 (Marca AND), equipo de disolución PT-DT7 (Pharma Test), espectrofotómetro UV-Visible V-630 (JASCO), potenciómetro 410 A (Orion), durómetro THT2 (BIOBASE), equipo de desintegración PTZ S (Pharma Test), bomba de vacío (Vacum Thomas), agitador magnético No: SP131320-33 (Thermo SCIENTIFIC, CIMAREC).
Pruebas de control de calidad
Antes de realizar los perfiles de disolución in vitro, los productos a evaluar se sometieron al control de calidad para formas farmacéuticas sólidas orales: Verificación de la integridad y rotulación del acondicionamiento, Características organolépticas, Peso promedio, Dureza y Desintegración, Pruebas de Identificación y Valoración del principio activo, Uniformidad de Unidades de Dosificación y Prueba de disolución, según la monografía de la monografía Monografía Clorhidrato de Metformina, Tabletas de la farmacopea USP 39/NF 34.
Elección de las condiciones del método de disolución para los perfiles de disolución
Tomando en cuenta que la División de BE recomienda que, para los PFMF, si hay un método de USP disponible para el producto, entonces la disolución se debe realizar utilizando ese método (Anand, Yu, Conner y Davit, 2011, párr. 5), se consideraron las condiciones establecidas en la monografía Clorhidrato de Metformina, Tabletas, de la USP 39/NF 34 (2016e, pp. 5244 y 5245), que considera 3 posibles métodos de disolución, de los cuales la Prueba 1 y la Prueba 3 emplean el Aparato 1 (canastillas), en las mismas condiciones, difiriendo en el tiempo de la prueba de 45 minutos y 60 minutos respectivamente. En la Prueba 2 se emplea el Aparato 2 (paletas). Para decidir cuál de los métodos se empleará para la realización de los perfiles de disolución en el estudio de BE in vitro, se probaron los ensayos tanto con el Aparato 1 (canstillas a 100 rpm) y con el Aparato 2 (paletas a 75 rpm), con 1000 ml de medio de disolución: solución amortiguadora de fosfato de pH 6,8 a 37 ± 0,5, tiempos de muestreo: 5 min, 15 min, 30 min, 45 min, 60 min y 75 min, tomando 10 ml de muestra sin reemplazo del medio de disolución, que son suficientes para obtener perfiles de disolución significativos, que cubren toda la disolución del principio activo metformina clorhidrato en los comprimidos de liberación inmediata, cumpliendo de esta manera la recomendación de la EMEA (2010) y la FDA (2017a), la lectura de las muestras se hizo en el espectrofotómetro UV-visible a 233 nm, con medio de disolución como blanco.
Selección de los lotes para el estudio de BE in vitro
Habiendo verificado que todos los lotes en estudio cumplen con los parámetros de calidad para comprimidos de metformina clorhidrato de liberación inmediata, se continuó con la selección de los lotes que entrarán al estudio de BE in vitro. Para ello, se realizaron los perfiles de disolución de cada lote en estudio, tomando 6 unidades por lote, con la técnica de la USP 39/NF 34, con el aparato 1, seleccionada. Se comprobó mediante el análisis estadístico ANOVA, que no exista diferencia significativa entre los perfiles de disolución de los lotes de cada producto en estudio. Se seleccionó el lote de cada producto que entrarán al estudio de BE in vitro, mediante el análisis cinético comparativo de liberación-disolución por el método de las pendientes.
Perfiles de disolución del Estudio de BE in vitro
Con el lote seleccionado de cada producto se realizaron los perfiles de disolución en 12 comprimidos de cada lote de acuerdo a la técnica seleccionada en base a la Monografía Clorhidrato de Metformina, Tabletas de la USP 39/NF 34, con seis tiempos de muestreo: 5, 15, 30, 45, 60 y 75 minutos. Se compararon los perfiles de disolución del fármaco metformina clorhidrato de los productos prueba y del producto de referencia, mediante el método independiente, con el cálculo de los factores de similitud (f1) y diferencia (f2), esperando establecer la BE in vitro entre los productos prueba y el producto referencia, obteniendo un valor de f2 entre 50 y 100 y un valor de f1 de 0 a 15, según los criterios de la FDA (1997, p. 8) y la OMS (2006), para poder respaldar la intercambiabilidad terapéutica entre estos productos.
RESULTADOS
Se escogieron 5 productos disponibles en el mercado boliviano con Registro Sanitario vigente: 4 PFMF y el producto de referencia Glucophage® (FDA, 2018), estos fueron seleccionados aleatoriamente en diferentes establecimientos farmacéuticos y regionales de distribución de la ciudad de La Paz en Bolivia. De cada producto se seleccionaron 3 lotes diferentes (A1, A2, A3; B1, B2, B3; C1, C2, C3; D1, D2, D3 y E1, E2, E3), con vencimiento vigente.
Pruebas de control de calidad
Los resultados obtenidos detodos los lotes en estudio cumplen con los requerimientos de calidad especificados en la monografía Monografía Clorhidrato de Metformina, Tabletas de la farmacopea USP 39/NF 34.
Elección de las condiciones del método de disolución para los perfiles de disolución
El principio activo metformina clorhidrato se libera más rápidamente del producto referencia A1, cuando se emplea las condiciones del método con el aparato 2, en comparación al método 1 con el Aparato 1, por tal motivo, para la realización de los perfiles de disolución en el estudio de BE in vitro, se tomaron en cuenta las condiciones del Método 1, que emplea el aparato 1, eligiendo de esta manera el método más crítico (el peor caso) para la liberación del principio activo metformina clorhidrato.
Selección de los lotes para el estudio de BE in vitro
De las 6 unidades por cada lote en estudio se obtuvieron los siguientes perfiles de disolución:

En las figuras se observan que los perfiles de disolución de A1, A2 y A3; C1, C2 y C3; D1, D2 y D3; E1, E2 y E3 son gráficamente similares, en cambio los perfiles de disolución de B1, B2 y B3 no son gráficamente similares. La significancia estadística de estas similitudes y diferencias halladas se verificaron con el análisis estadístico ANOVA (método FISHER). Por esta razón, el producto B se eliminó del estudio, quedando los lotes de los productos A, C, D y E para el análisis cinético, para determinar la constante de velocidad de disolución (kd) intermedia de cada producto (min-1) y de esta manera escoger los lotes que entraron al estudio de BE in vitro, obteniendo los siguientes resultados de Kd promedio:
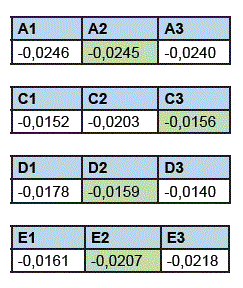
En función a los resultados se seleccionaron los lotes con el valor intermedio de kd para el estudio de BE in vitro: el lote “A2” como el lote de referencia y los lotes “C3”, “D2” y “E2” de los productos prueba; también, se corroboró que las potencias de estos últimos lotes no difieren en más del 5%, de la potencia del lote del producto de referencia seleccionado (A2).
Perfiles de disolución del Estudio de BE in vitro
Los coeficientes de variación de los PDA de los productos A2, C3, D2 y E2 en los tiempos de muestreo más tempranos (5 y 15 minutos) fueron menores a 20% y para los demás tiempos de muestreo (30, 45, 60, y 75 minutos) se obtuvieron valores menores del 10%, lo cual permitió utilizar el dato promedio del PDA por cada tiempo de muestreo de los 12 comprimidos, para el análisis de comparación de los perfiles de disolución mediante el método de modelo independiente, cumpliendo las recomendaciones de la OMS (2006) y de la FDA (2017a).
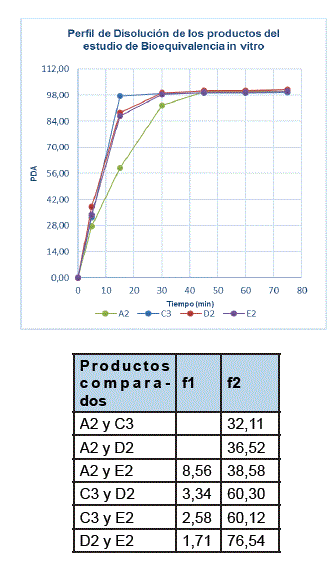
A continuación, se muestran los resultados obtenidos de los perfiles de disolución del estudio de BE in vitro de los productos A, C, D y E; y los resultados del factor de diferencia (f1) y del factor de similitud (f2):
DISCUSIÓN
A los 30 minutos el porcentaje de disolución alcanzado de los 3 lotes del producto A (referencia), de los 3 lotes del producto D y de los 3 lotes del producto E fue mayor a 85%, por lo que se consideran de rápida disolución en la Solución amortiguadora de fosfato de pH 6,8, según la FDA (2017a), OMS (2006) y la EMEA (2010). A los 15 minutos el porcentaje de disolución alcanzado de los 3 lotes del producto C, fue mayor a 85%, por lo que puede considerarse de muy rápida disolución en la Solución amortiguadora de fosfato de pH 6,8, según la FDA (2017a), OMS (2006) y la EMEA (2010). El porcentaje de disolución alcanzado a los 5 minutos, de los lotes B2 y B3 fue mayor al 85%, por lo que podrían considerarse de muy rápida disolución en la Solución amortiguadora de fosfato de pH 6,8, pero los resultados del lote B1 muestran que la liberación del principio activo no tiene comportamiento similar a los lotes B2 y B3, observándose una disolución lenta del principio activo, que alcanza más del 85% recién a los 45 minutos. Esta variación en la liberación del principio activo metformina clorhidrato, probablemente se deba a que el proceso de fabricación del producto B, no es reproducible entre lotes. Por lo tanto, se puede inferir que no se han validado los procesos de fabricación de este producto, no cumpliendo con las normas de Buenas Prácticas de Manufactura, o que los comprimidos del lote B1 fueron afectados por las condiciones de temperatura y humedad ambientales, a raíz de un mal almacenamiento. La rápida velocidad de disolución de los lotes B2 y B3, en comparación a los demás productos en estudio (A, C, D y E), podría deberse a que estos probablemente contengan una mayor cantidad de desintegrante en su composición.
Al comparar los perfiles de disolución de los PFMF y del producto de referencia, se presenta contradicción entre los resultados de f1 y f2, por lo que la decisión prima sobre el factor de similitud (f2), cuyos resultados obtenidos son menores a 50, según la OMS (2006) y la FDA (2017a), esto quiere decir que no existe equivalencia del desempeño in vitro entre los productos PFMF en estudio (C, E y D) y el producto de referencia Glucophage® (A), por lo tanto, en esta investigación se demuestra que no son bioequivalentes in vitro. Al comparar los perfiles de disolución de los productos PFMF (C, D y E) entre sí, utilizando el enfoque matemático del modelo independiente, se obtuvieron valores de f1 que están dentro del intervalo de 0 a 15 y valores de f2 dentro del intervalo de 50 a 100, demostrándose de esta manera que existe equivalencia del desempeño in vitro entre los PFMF C, D y E.
CONCLUSIONES
En el presente estudio se ha visto cuán importante es poder demostrar la BE de los PFMF con el producto innovador o de referencia, para demostrar que ambos son terapéuticamente equivalentes y que en la práctica clínica puedan ser intercambiados unos con otros, respaldando de esta manera la calidad, eficacia y seguridad de los PFMF.
Con el presente estudio de bioexención en base al SCB, se constató que los estudios de BE in vitro son una herramienta útil y económica en comparación a los estudios de BE in vivo, para establecer la intercambiabilidad de los productos farmacéuticos y respaldar la calidad, seguridad y eficacia de los PFMF.
De los 4 PFMF seleccionados para determinar la BE in vitro, el producto B (importado) fue eliminado de este estudio, por no cumplir el requisito de uniformidad de lotes, evaluado por el método estadístico ANOVA, quedando 3 PFMF de fabricación nacional.
De los 3 PFMF se seleccionaron los lotes C3, D2 y E2, para el estudio de BE in vitro, y el lote A2 del producto referencia, por medio del análisis cinético comparativo de liberación-disolución por el método de las pendientes, a través de la velocidad de disolución intermedia.
Los 3 PFMF de liberación inmediata, de administración peroral, a base de metformina clorhidrato, de procedencia nacional que entraron al estudio de BE, no demostraron BE in vitro frente al producto de referencia, desde el punto de vista de la velocidad de disolución y porque los valores hallados de f2 (factor de similitud) fueron menores a 50. Sin embargo, estos 3 PFMF se consideran biodisponibles, porque el producto C, es de muy rápida disolución (más del 85% de la cantidad declarada de metformina clorhidrato en el comprimido, se disuelve en 15 minutos), además, los productos D y E son de rápida disolución (más del 85% de la cantidad declarada de metformina clorhidrato en el comprimido, se disuelve en 30 minutos), lo que respaldaría su eficacia, Así mismo, estos PFMF son equivalentes farmacéuticos al de referencia, porque contienen la misma cantidad de IFA, en la misma forma farmacéutica, con la misma ruta de administración y reúnen estándares convencionales de potencia, calidad, pureza e identidad,
La OMS en su Anexo 7 del Informe 40 indica, que la evidencia del comportamiento in vitro encontrado, podría pasar desapercibido en el comportamiento in vivo.
Los PFMF pertenecen a la Clase 3 y su Bioexención basada en el SCB, para la BE debe estar científicamente justificada. Según la FDA los productos prueba deben contener los mismos excipientes del producto de referencia. La composición de estos productos debe ser cualitativamente la misma del producto de referencia (excepto por los colorantes, saborizantes o preservantes) y cuantitativamente muy similar al mismo, debido a que los excipientes podrían influir en la liberación del ingrediente farmacéutico activo, afectando su Biodisponibilidad. Por otro lado, la FDA también pide incluir el método de fabricación de los productos prueba, porque puede influir en la velocidad de liberación del ingrediente farmacéutico activo.
En el presente trabajo y para cualquier otro similar, en el que se determine la bioexención del ingrediente farmacéutico activo de Clase 3, es difícil llegar a obtener la composición cuali-cuantitativa tanto de los productos prueba, como del producto de referencia, porque no se tiene acceso a esta información, que es propia de cada fabricante.
Se verificó el cumplimiento de las especificaciones de calidad de los 4 PFMF evaluados según farmacopea USP 39/NF 34.
Al comparar los perfiles de disolución de los PFMF entre sí (C3, D2 y E2), utilizando el enfoque matemático del modelo independiente se demostró que existe equivalencia del desempeño in vitro entre los mismos.
REFERENCIAS BIBILIOGRAFÍCAS
Anand, O., Yu, L.X., Conner, D.P. y Davit, B. M. (2011), Dissolution Testing for Generic Drugs: An FDA Perspective. The AAPS Journal. 13 (3). Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3160163/. [Consultado el 14 de abril de 2018]. [ Links ]
Baena, Y y Ponce D`León, L.F. (2004). Importancia y fundamentación del sistema de clasificación biofarmacéutico, como base de la exención de estudios de Biodisponibilidad y Bioequivalencia in vivo. Revista Colombiana de ciencias Químicas y Farmacéuticas. 33 (1). pp. 18-32. Recuperado de: http://www.scielo.org.co/pdf/rccqf/v37n1/v37n1a02.pdf. [ Links ]
European Medicines Agency (EMEA), (2010). Guideline on the investigation of bioequivalence. Recuperado de: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. [Consultado el 20 de abril de 2018]. [ Links ]
FDA Center for Drug Evaluation and Research (CDER). (1997). Guidance for Industry - Dissolution testing of immediate release solid oral dosage forms. Recuperado de: https://www.fda.gov/downloads/drugs/guidances/ucm070237.pdf. [Consultado el 13 de abril de 2018]. [ Links ]
FDA Center for Drug Evaluation and Research (CDER). (2017a). Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate Release Solid oral dosage Forms based on a Biopharmaceutic Classification System. Recuperado de: https://www.fda.gov/downloads/Drugs/Guidances/ucm070246.pdf. [Consultado el 13 de abril de 2018]. [ Links ]
FDA. (2018). Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations. Recuperado de: https://www.accessdata.fda.gov/scripts/cder/ob/search_product.cfm [ Links ]
Honorable Congreso Nacional de Bolivia. (1996). Artículo 1. Ley del Medicamento Nª 1737 del Estado Plurinacional de Bolivia. Recuperado de: http://agemed.minsalud.gob.bo/reg-far/1.htm.[Consultado el 03 de abril de 2017]. pp.1–4. [ Links ]
Instituto Nacional de Estadística (INE). (14 de noviembre de 2017). En 2016 se registraron 138.124 casos de diabetes. Recuperado de: https://www.ine.gob.bo/index.php/principales-indicadores/item/2203-en-2016-se-registraron-138-124-casos-de-diabetes. [ Links ]
Malinowski, H. J. (2003). Biodisponibilidad y pruebas de Bioequivalencia. En Gennaro, A.R. Remington Farmacia. pp. 1-2506. Buenos Aires - Argentina: Panamericana. [ Links ]
Makoid, M. C., Vuchetich, P. J. y Banakar, U. V. (1999). Basic Pharmacokinetics, Nebraska. [ Links ]
Midha, K. y McKay, G. (2009). Bioequivalence; Its History, Practice, and Future. The A.A.P.S. Journal. 11 (4), pp. 664-670. doi: 10.1208/s12248-009-9142-z. [ Links ]
Ministerio de Salud del Estado Plurinacional de Bolivia. (2016). Listado de medicamentos Esenciales (LINAME). Recuperado de: https://www.minsalud.gob.bo/contactos/liname-2014-2016. [ Links ]
Ministerio de Salud del Estado Plurinacional de Bolivia. (26 de septiembre de 2017). En Bolivia se incrementa casos de Diabetes Mellitus y el Ministerio de Salud busca mejorar diagnóstico de la enfermedad. Recuperado de: https://www.minsalud.gob.bo/2703-en-bolivia-se-incrementa-casos-de-diabetes-mellitus-y-el-ministerio-de-salud-busca-mejorar-diagnostico-de-la-enfermedad-2. [Consultado el 12 de mayol de 2018]. [ Links ]
Ministerio de Salud de la República de Argentina. sin fecha. Diabetes. Disponible en: http://www.msal.gob.ar/ent/index.php/informacion-para-ciudadanos/diabetes. [Consultado el 12 de mayol de 2018]. [ Links ]
Placencia, M. (2010). La Bioequivalencia como requisito de calidad de los medicamentos genéricos/multifuente: estudio comparativo en países latinoamericanos (Tesis de Doctorado). Facultad de Farmacia y bioquímica de la Universidad Nacional Mayor de San Marcos. Perú. Disponible en: http://www.cybertesis.edu.pe/. Lima–Perú. 03 de julio de 2013. pp. 4, 17, 18. [ Links ]
Ponce D`León, L.F. y Jaramillo, A. (2004). Estudio de Bioequivalencia in vitro de cuatro productos de amoxicilina del mercado colombiano. Revista Colombiana de Ciencias Químicas y Farmacéuticas. 33 (1). pp. 70-76. Recuperado de: https://revistas.unal.edu.co/index.php/rccquifa/article/view/1664/2322. [ Links ]
USP 39/NF 34 Farmacopea de los Estados Unidos de América – Formulario nacional. (2016e). Clorhidrato de Metformina, Tabletas. Volumen III. Estados Unidos de América. pp. 5244, 5245. [ Links ]
World Health Organization. (2006). Fortieth Report. Geneva. pp. 1-461. Recuperado de: http://apps.who.int/medicinedocs/documents/s14091e/s14091e.pdf. [Consultado el 14 de abril de 2018]. [ Links ]
World Health Organization. (2005a). Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability. Recuperado de: http://www.who.int/medicines/services/expertcommittees/pharmprep/QAS04_093Rev4_final.pdf. [Consultado el 14 de abril de 2018]. [ Links ]

