











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkRevista CON-CIENCIA
versión impresa ISSN 2310-0265
Rev.Cs.Farm. y Bioq vol.5 no.1 La Paz jun. 2017
ARTÍCULOS ORIGINALES
Estudio de propiedades moleculares de cuatro flavonoides de Baccharis boliviensis
Study of molecular properties of four flavonoids of baccharis boliviensis
CHOQUE, RIGOBERTO1 GEMIO, RÓMULO1 NOGALES, JORGE 2*
1 Carrera de ciencias químicas.FCPN-UMSA Cota Cota calle 27 campus universitario La Paz - Bolivia
2*Quimica General e Inorganica,carrera Quimica Farmaceutica.FCFB-UMSA.Av.Saavedra 2224 La Paz - Bolivia 2* Autor principal: jorge.nogales.vera@gmail.com
FECHA DE RECEPCIÓN: 23 ENERO DE 2017 FECHA DE ACEPTACIÓN: 27 DE ABRIL DE 2017
Resumen
En este trabajo de investigación se realizó un estudio de relación estructura y actividad antioxidante empleando métodos computacionales basándose en propiedades estructurales y electrónicas, para lograr este objetivo se utilizó el programa computacional Hyperchem versión 8.0.1 empleando métodos cuánticos semiempiricos, a partir de las estructuras más estables obtenidas se calcularon longitudes de enlace, cargas atómicas, entalpias de disociación de enlace y orbitales moleculares de los cuatro flavonoides de Baccharis boliviensis aislados e identificados por el grupo de investigadores de Almanza, G Et.al en el año 2012. Al relacionar todos los datos obtenidos con la capacidad antioxidante se encontró que la actividad antioxidante entre los cuatro flavonoides el flavonoide 4 tiene mayor capacidad antioxidante debido al mayor número de grupos hidroxilo y a los valores bajos de energía de disociación de enlace EDE relativa respecto al flavonoide 1 que ocuparía el segundo lugar en términos de capacidad antioxidante, apoyan a estos resultados el hecho de considerar los valores de la segunda diferencia de energía de disociación de enlace EDE entre estos dos flavonoides se obtiene una diferencia de 1.106 H (Hartrees) para el flavonoide 4 y 1.4651H para el flavonoide 1. Los flavonoides 2 y 3 ocuparían las posiciones 3 y 4 en términos de capacidad antioxidante respectivamente. En conclusión la actividad antioxidante de los cuatro flavonoides estudiados presenta el siguiente orden de mayor a menor actividad antioxidante: flavonoide 4 > flavonoide 1 > flavonoide 2 > flavonoide 3
PALABRAS CLAVE
Antioxidante, métodos semiempíricos, Hyperchem, Baccharis boliviensis, química computacional
Abstract
In this research work was carried out a study of relationship structure and antioxidant activity using computational methods based on structural and electronic properties, to achieve this objective we used the computer program Hyperchem version 8.0.1 using semiempirical quantum methods, from structures More stable, bond lengths, atomic charges, bond dissociation enthalpies and molecular orbitals of the four Baccharis boliviensis flavonoids isolated and identified by the Almanza group, G Et.al in 2012, were calculated. By relating all the data obtained with the antioxidant capacity it was found that the antioxidant activity among the four flavonoids f lavonoid 4 has higher antioxidant capacity due to the higher number of hydroxyl groups and the lower energy values of linkage dissociation EDE relative to flavonoid 1 thatwould occupy the second place in terms of antioxidant capacity, support to these results the fact of considering the values of the second energy difference of linkage dissociation EDE between these two flavonoids gives a difference of 1,106 H (Hartrees) for the flavonoid 4 and 1.4651 H for flavonoid 1.The flavonoids 2 and 3 would occupy positions 3 and 4 in terms of antioxidant capacity respectively. In conclusion the antioxidant activityof the four flavonoids studied has the following order of highest to lowest antioxidantactivity: Flavonoid 4> flavonoid 1 > flavonoid 2> flavonoid 3
KEY WORDS
Antioxidant, semiempirical methods, Hyperchem, Baccharis boliviensis, computational chemistry
INTRODUCCIÓN
Los flavonoides son pigmentos naturales presentes en los vegetales y que protegen al organismo del daño producido por agentes oxidantes, como los rayos ultravioleta, la polución ambiental, sustancias químicas presentes en los alimentos y otros; desempeñan un papel esencial en la protección frente a los fenómenos de daño oxidativo, y tienen efectos terapéuticos en un elevado número de patologías, incluyendo la cardiopatía isquémica, la aterosclerosis y el cáncer.(Allen,2000:463). Sus propiedades anti-radicales libres se dirigen fundamentalmente hacia los radicales hidroxilo y superóxido, especies altamente reactivas implicadas en el inicio de la cadena de peroxidación lipídica y se ha descrito su capacidad de modificar la síntesis de eicosanoides con respuestas antiinflamatoria. (Sies,1986:1058).
Según Allen,(2000:464) el organismo humano no puede producir estas sustancias químicas protectoras, por lo que deben obtenerse mediante la alimentación o en forma de suplementos. Están ampliamente distribuidos en plantas, frutas, verduras y en diversas bebidas y representan componentes sustanciales de la parte no energética de la dieta humana.
Estos compuestos fueron descubiertos por el premio Nobel Szent-György, quien en 1930 aisló de la cáscara del limón una sustancia, la citrina, que regulaba la permeabilidad de los capilares. Los flavonoides se denominaron en un principio vitamina P (por permeabilidad) y también vitamina C2 (porque se comprobó que algunos flavonoides tenían propiedades similares a la vitamina C). Sin embargo, el hecho de que los flavonoides fueran vitaminas no pudo ser confirmado, y ambas denominaciones se abandonaron alrededor de 1950. Los flavonoides contienen en su estructura química un número variable de grupos hidroxilo fenólicosy excelentes propiedades de quelación del hierro y otros metales de transición, lo que les confiere una gran capacidad antioxidante.(Rybczynska,1994:419)
El año 2012 fueron reportados el aislamiento y elucidación estructural de cuatro flavonoides no reportados anteriormente en Baccharis boliviensis, identificados como: flavonoide 1 (5,7,3',4'-tetrahidroxi-6-metoxiflavonona), flavonoide 2 (5,3'-dihidroxi-3,6,7,8,4'-pentametoxiflavona), flavonoide 3 (5,4'-dihidroxi-6,7,8-trimetoxiflavona) y flavonoide 4 (5,6,7,3',4'-pentahidroxi-flavona), por el grupo de investigación de Almanza, G et.al en la Revista Boliviana de Química. (Almanza G, 2012:155)
No existe hasta la fecha trabajos sobre estas moléculas que describan sus propiedades moleculares desde el punto de vista computacional.
El presente trabajo se enfoca principalmente en estos compuestos y tiene como objetivo principal estudiar la reactividad de estos cuatro flavonoides con la finalidad de establecer las propiedades estructurales y electrónicas que influyen y afectan a dicha reactividad.
Los estudios realizados a todas las estructuras son en su totalidad teóricos y las propiedades evaluadas fueron: Geometría Estructural (arreglo espacial de la molécula, ángulos, longitudes de enlace), distribución de carga (que átomos tienen caracter positivo o negativo ), entalpia de disociación de enlace O-H, densidad electrónica a través del estudio de los orbitales HOMO y LUMO.
QUÍMICA COMPUTACIONAL
Históricamente, la Química Computacional surge como una evolución de la Química Teórica con el objetivo de investigar el comportamiento de la materia a nivel molecular mediante ordenadores (Sanchez R,2000:80); en otras palabras, la Química Computacional es sinónimo de modelización molecular. Más concretamente, la implementación y aplicación de las técnicas de la Química Computacional nos permite investigar múltiples propiedades y comportamientos moleculares como son:
- La geometría molecular en un sentido amplio, además de distancias y ángulos de enlace, es posible caracterizar la forma y tamaños relativos de todo tipo de moléculas y macromoléculas.
- La energía de todo tipo de especies químicas, incluyendo intermedios, estados de transición, estados excitados, etc. Es posible, además, estimar distintos tipos de magnitudes termodinámicas tanto en fase gas como en fases condensadas.
- La reactividad química de una especie es un ejemplo de propiedad genuinamente química que puede cuantificarse en forma de índices de reactividad con la ayuda de la Química Computacional.
Además, la Química Computacional es capaz de determinar la geometría y estabilidad relativa de intermedios y estados de transición de reacción, es decir, caracteriza los mecanismos de reacción. Más aún dispone de herramientas sofisticadas para el cálculo de todo tipo de magnitudes cinéticas como constantes de velocidad, efectos cinéticos isotópicos, secciones eficaces de reacción, factores de efecto túnel, etc. (Sanchez R,2000:81)
Métodos de la Mecánica Molecular (MM)
Estos métodos estiman el cambio en la energía potencial de un sistema molecular como consecuencia de pequeñas variaciones en las distancias y ángulos de enlace y, sobre todo, de los cambios conformacionales y la formación/ruptura de interacciones intermoleculares. Su formulación práctica requiere la adopción a priori de potenciales de interacción que contienen parámetros optimizados a partir de información experimental o teórica. Su gran ventaja reside en la rapidez de los cálculos MM, lo que permite modelizar sistemas con miles y cientos de miles de átomos. Mediante la aplicación de condiciones periódicas, los métodos MM nos permiten simular exhaustivamente el comportamiento dinámico y las propiedades termodinámicas de las fases condensadas. Sus principales limitaciones son que no proporcionan ninguna información o propiedad electrónica y su ámbito de aplicación viene predeterminado por los potenciales y parámetros empleados. (Suarez D,2000:23)
Muchos autores arguyen que la mecánica molecular no es más que un método de ajuste del problema a la idealidad, no de cálculo molecular exactamente. En estos métodos, la energía se deriva de una serie de contribuciones individuales como la energía de tensión, de torsión, de flexión, de repulsión e interacciones no enlazantes.
Todos los parámetros que se emplean en mecánica molecular son calculados por medio de métodos cuánticos o con métodos experimentales, y se denomina campo de fuerzas. Según la modelización que se quiera hacer, existen diversos campos de fuerza, unos más genéricos que otros. Por ejemplo, el OPLS fue desarrollado para ácidos nucleicos y proteínas y cuenta con un tratamiento de las interacciones no enlazantes más exacto que el AMBER, otro campo de fuerzas algo más antiguo. En este tema no hay que poseer unos conocimientos matemáticos desorbitados para poder desarrollar un campo de fuerzas, por lo que hay bastantes campos desarrollados por las mismas empresas que comercializan los programas, aunque los campos clásicos cuentan con más prestigio, al estar ampliamente reconocidos durante bastantes años. (Suarez D,2000:27).
Métodos Semiempíricos (SE)
Los métodos semiempíricos (SE) son métodos químico cuánticos, es decir, obtienen la energía y orbitales moleculares, pero lo hacen simplificando los cálculos HF o DFT al remplazar valores esperados de la función de onda o de funcionales de la densidad electrónica por formas paramétricas ajustadas estadísticamente empleando grandes conjuntos de datos experimentales (geometrías moleculares y calores de formación) y/o datos teóricos generados por métodos ab initio o DFT. (Suarez D,2000:26)
De esta manera, los cálculos SE son casi tan rápidos como los cálculos MM hasta el punto que es posible realizar cálculos SE en sistemas de miles de átomos. Por su naturaleza, la fiabilidad de los métodos SE es baja si se estudian moléculas poco representativas (por ejemplo, con metales de transición) y/o propiedades moleculares que no se han tenido en cuenta en la parametrización. (Suarez D,2000:27)
Las desventajas que presentan los métodos puramente cuánticos hacen que su uso en sistemas factibles a pie de laboratorio sea muchas veces descabellado, tanto por el tiempo como por la potencia del ordenador requerido. Es por ello que para poder explicar de manera teórica algunos resultados experimentales se requiere únicamente exactitud química, es decir, un margen de error de algunas Kcal/mol, no de 10e-12 eV como puede ser en aplicaciones teóricas. Para ello, lo primero que se planteó fue el simplificar los métodos cuánticos allí donde se pudiera hacer, surgiendo de esta forma los métodos semiempíricos. (Suarez D,2000:29)
A un nivel fundamental, estos métodos simplifican las integrales que aparecen durante el cálculo, haciendo cero algunas y sustituyendo otras por funciones analíticas en las que aparecen parámetros, estos parámetros se obtienen para cada átomo en cuestión por medio de procedimientos experimentales ó bien por medio de cálculos cuánticos de alto nivel, el resultado es que, aparte de incluir efectos reales que los abinitio tendrían que calcular o aproximar, el ahorro de tiempo y de almacenamiento de integrales desciende dramáticamente hasta un 50% ó más. (Suarez D,2000:30)
Otros problemas relativos al manejo de integrales también quedan eliminados al sustituir la mayoría de éstas por expresiones fácilmente resolubles en el ordenador. Los métodos más usados a la hora de la verdad suelen ser el AM1 y el PM3, desarrollados por el profesor Dewar, aunque aún perviven otros más antiguos como el CNDO, el INDO y el MINDO/3. Todos ellos dan resultados satisfactorios cuando se trata de obtener calores de formación, energías de conformaciones e incluso análisis de reacciones químicas, siempre con cuidado en este último caso, ya que los parámetros que emplean estos programas son para átomos en su estado fundamental, no en un estado excitado o en un estado de transición.
Tanto los métodos puramente cuánticos como los semiempíricos comparten un problema que, si bien no es tanto en un laboratorio de química normal, sí que lo es cuando entramos en uno de bioquímica: El tamaño de las biomoléculas (varios miles de átomos) hace que un abinitio se colapse por el propio algoritmo de integración o de cálculo ó que un semiempírico alcance el infinito en tiempo de cálculo o se colapse también. El desarrollo de procedimientos aún menos costosos fue necesario para poder comenzar a estudiar el comportamiento de las macromoléculas en disolución y de su interacción con ligandos como fármacos o inhibidores. La respuesta a esta solicitud fueron los métodos de mecánica molecular. En ellos, la única teoría que se emplea es la mecánica clásica, ya que trata a los átomos como bolas de una masa determinada y a los enlaces como muelles. Al tener que resolver ecuaciones muchísimo más simples, el ahorro de tiempo es aún mayor que en los semiempíricos, posiblemente un 70-80%, sin contar que no almacenan integrales. (Suarez D,2000:32)
METODOLOGÍA
El estudio de modelado molecular se desarrolló utilizando el programa Hyperchem 8.0.1 empleando un procesador Intel core i7 en un entorno windows. Las estructuras utilizadas fueron los cuatro flavonoides de Baccharis boliviensis reportados en La Paz Bolivia por el grupo de investigación de Almanza, G. Et.al., publicados en la Revista Boliviana de Química VOL. 29, No.2 del año 2012.
Estos flavonoides y las estructuras de todos los posibles radicales de cada flavonoide fueron trazadas usando la interfaz de Hyperchem 8.0.1, todas las estructuras fueron sometidas a cálculos de optimización geométrica inicialmente de Mecánica Molecular para obtener la estructura más estable, posteriormente todas las estructuras fueron analizadas mediante el método semiempirico AM1 para obtener valores más confiables de energía y geometría.
Las geometrías moleculares optimizadas fueron consideradas como la conformación con el mínimo de energía porque los modos vibracionales fueron calculados al mismo nivel de teoría y se observa que todas las frecuencias de las estructuras fueron positivas (números reales) por tanto se confirma que corresponden a mínimos verdaderos en la superficie de energía potencial SEP del sistema. La minimización de la energía de las estructuras se desarrolló por el método Fletcher-Reeves de gradiente conjugado hasta un valor de gradiente RMS (root-mean-square) menor a 0.100 Kcal/(A°mol) o un máximo de 500 ciclos de iteración, el límite de convergencia SCF empleado fue de 0.001 Kcal/mol.
Las energías de los orbitales HOMO y LUMO medidos en (ev) de cada molécula fueron calculados una vez terminada la optimización geométrica aplicando los algoritmos del Hyperchem. Los mapeos moleculares de los potenciales electrostáticos se obtuvieron usando la opción plot molecular properties del menú compute de este programa y posteriormente fueron analizados. Todos los datos obtenidos se almacenan en un archivo de salida para cada molécula de flavonoide y sus radicales denominado archivo log que contiene cada calculo aplicado; las estructuras se guardan con extensión.hin. El ordenamiento de los datos numéricos se realizaron en Excel 2015.
RESULTADOS Y DISCUSIÓN
Se calcularon las estructuras de los cuatro flavonoides utilizando de manera inicial la mecánica molecular con lo que se obtuvieron las estructuras más estables para cada flavonoide, posteriormente estas estructuras se sometieron a cálculos utilizando métodos semiempiricos para obtener valores más confiables de energía y geometría.
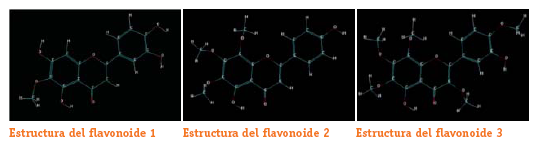
Estas estructuras corresponden a las optimizadas por método semiempirico de los cuatro flavonoides de Baccharis boliviensis.
Se muestran las estructuras de menor energía caracterizadas como mínimos verdaderos. Al igual que para las estructuras de los radicales que se mostraran posteriormente, los modos vibracionales fueron calculados por el programa y se observa que todas las frecuencias de las estructuras fueron positivas (números reales) por lo tanto se confirma que corresponden a mínimos verdaderos en la superficie de energía potencial SEP.
Geometría molecular
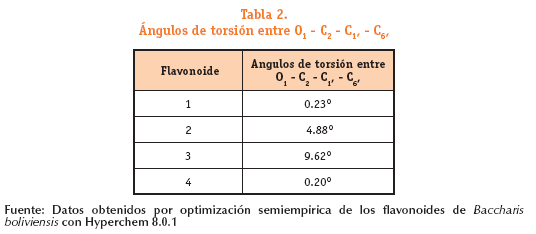
Los ángulos de torsión entre O1 - C2 - C1, - C6, son: 0,23° para el flavonoi-de 1; 4,88° para el flavonoide 2; 9,62° para el flavonoide 3; y 0,20° para el fla-vonoide 4.
Estos resultados obtenidos en cuanto a la optimización geométrica indican que los cuatro flavonoides presentan una geometría molecular plana.
Análisis conformacional de la geometría molecular por optimización
Durante el cálculo de la energía total de los flavonoides analizados por métodos computacionales en la optimización geométrica se encontraron valores que permiten plantearse una idea acerca de la reactividad
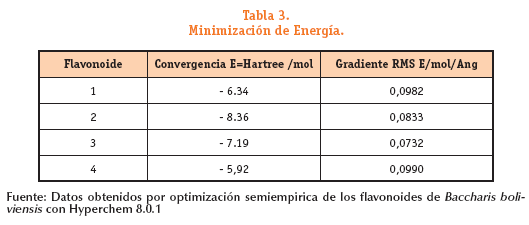
Esta tabla nos permite proponer que las moléculas con los menores valores de energía total durante la optimización geométrica son el flavonoide 2 y el flavonoide 3, siendo catalogados como moléculas con la estructura más estable y por lo tanto con la menor reactividad. Mientras que las moléculas con la mayor reactividad deben ser el flavonoide 4 y el flavonoide 1, de acuerdo con los valores encontrados de energía. El gradiente RMS durante el análisis de datos brinda la información del comportamiento de los enlaces químicos y su efecto sobre la conformación más estable.
El flavonoide 3 y el flavonoide 2 poseen los gradientes de energía menores en comparación con los otros, evidenciando su estabilidad Al observar esta misma tabla se ve que las energías de los flavonoides 1 y flavonoide 4 presentan valores muy cercanos apareciendo en primer lugar el flavonoide 4 y luego el flavonoide 1 de acuerdo al criterio del valor de gradiente RMS. De acuerdo con los datos reportados en la tabla N°3 es posible plantear un orden de reacción en función a la reactividad respecto a los valores de energía mínima total al finalizar la optimización de la geometría molecular: flavonoide 4; flavonoide 1; flavonoide 3 y por ultimo el flavonoide 2. Distribución de cargas en los flavonoides (ver tabla 4)
La distribución de cargas fue calculada en términos de cargas naturales de Lewis. Tabla 4 y Figura 5. El valor más negativo en cada estructura pertenece a los oxígenos del C3, del flavonoide 1 (-0.268); C3, del flavonoide 2 (-0.262); C4, del flavonoide 3 (-0.243) y C5 del flavonoide 4 (-0.273) que posteriormente demostraremos que es el C6 quien forma el radical más estable al observar el valor más negativo para su respectivo radical. Estas cargas negativas correspondientes a los oxígenos proporcionan una primera idea del lugar donde se formara el radical más estable de cada estructura.
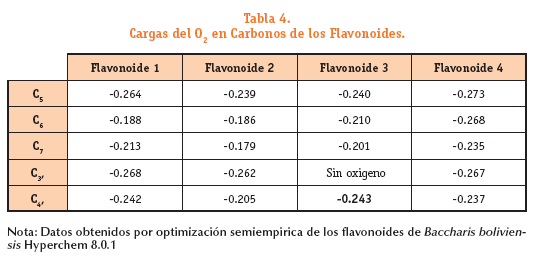
Distribución de cargas en los flavonoides y sus radicales:
En la tabla podemos ver los valores de carga del oxígeno (hidroxilo) de las estructuras primarias de los flavonoides y de sus respectivos radicales donde se perdió el hidrogeno del hidroxilo.
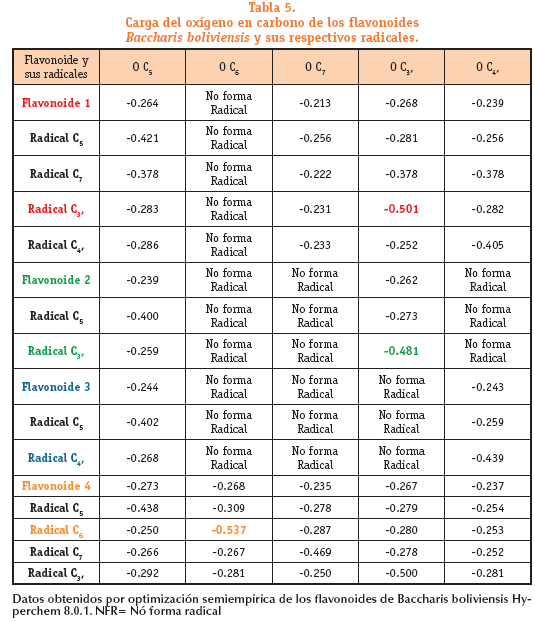
El valor más negativo en cada estructura (ver tabla 5) pertenece a los oxígenos del C3, del radical C3, del flavonoide 1 (-0.501); C3, del radical C3, del flavonoide 2 (-0.481); C4, del radical C4, del flavonoide 3 (-0.439) y C6 del radical C6 del flavonoide 4 (-0.537). Estas cargas negativas correspondientes a los oxígenos confirman el lugar donde se forman los radicales más estables en cada estructura. Esta predicción es correcta para todas las estructuras.
Análisis electrónico
Perez,G. y col (2003) reportan que existe una relación directa entre el número de hidroxilos y la capacidad antioxidante. Otros autores han concluido que la actividad antioxidante en flavonoides no solo está relacionada con el número de hidroxilos, sino también con factores como el grado de deslocalización electrónica (acortamiento del enlace C2 - C1,) entre los anillos A,B y C, el número de hidroxilos en toda la estructura y por último la energía de disociación de enlace EDE. Para realizar el análisis electrónico se generaron las estructuras de los radicales a partir de las estructuras primarias de los cuatro flavonoides de Baccharis boliviensis se calcularon sus propiedades electrónicas para determinar cuál de los radicales formados para cada flavonoide es el más estable.
Diferencias de energías entre estructuras primarias y sus radicales.
En la Tabla 6 se muestran las energías calculadas por Hyperchem 8.0.1 para cada radical generado a partir de su estructura primaria.La energía esta expresada en Hartrees.Las diferencias de energía AE obtenidas para cada flavonoide se muestran en la tabla 6 y su valor se calcula restando la energía del radical a la energía de la estructura primaria. Esta diferencia permite predecir cuál es el radical más estable para cada compuesto.
En el flavonoide 1, la estructura del radical libre localizado en el grupo hidroxilo unido al C5 tiene una energía de 1006.788133 Hartrees este valor se resta al valor de la energía calculada para la molécula primaria que es de 1022.216229 Hartrees, la diferencia de energía es AE = 15.4281 Hartrees que en este caso no es el radical más estable, para el flavonoide 1 el radical con menor energía corresponde al grupo OH en la posición C3, siendo este el radical que presenta mayor estabilidad.
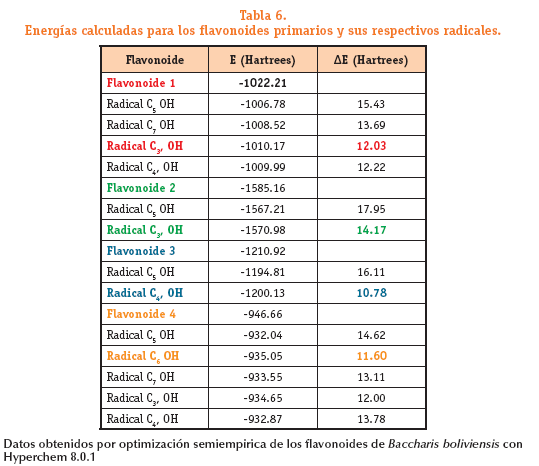
Este proceso de hallar las diferencias se realiza con todos los radicales de cada flavonoide. Para el flavonoide 2 se observa que las diferencias de energías más bajas se encuentran cuando el hidrogeno eliminado se encuentra unido al oxigeno del C3, con una AE = 14.1756 Hartrees. Los radicales más estables para el flavonoide 3 corresponde a las estructuras donde el hidrogeno eliminado se encuentra unido al oxigeno del C4, AE = 10.7863 Hartrees y C6 en la 6-Hidroxiluteolin AE = 11.6079 Hartrees. De acuerdo con los datos reportados en la tabla 6 es posible plantear el lugar donde se formara el radical más estable de acuerdo a los valores de AE:
El radical más estable cuando el hidrogeno liberado unido al oxígeno en posición de Carbono es C3, en el flavonoide 1; C3, en flavonoide 2; C4, en el flavonoide 3; y C6 en flavonoide 4.
Cálculo de las entalpias de disociación de enlace EDE
Se calcularon las entalpias de disociación de enlace EDE teniendo en cuenta la reacción:

El valor de la entalpía de disociación de enlace EDE para el átomo de hidrogeno (H *) calculado al mismo nivel de teoría que los flavonoides y sus radicales es de - 0,50 Hartrees.
Entalpia de Disociación de Enlace Relativa: EDE relativa
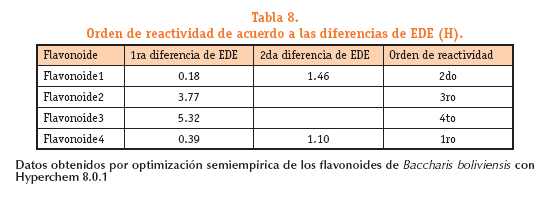
Los valores relativos de entalpia de disociación de enlace, se calcularon para cada flavonoide para ilustrar lo que ocurre a nivel estructural. El valor de EDE relativa se ha determinado para los radicales de cada flavonoide como la diferencia entre el menor valor de EDE y los otros valores de EDE de los demás radicales de la misma estructura del flavonoide.En la figura 7 se muestran con las flechas los sitios de ataque de las especies reactivas de oxigeno (EROs).
En el flavonoide 1 el enlace O-H unido al C3, es el sitio más probable de ataque porque tiene el valor de entalpia menor de disociación de enlace tabla N°7 (11.5382 Hartree) por lo que se le asigna el valor de 0.0 H ver figura 7; el grupo hidroxilo unido al C4, es el segundo sitio probable con un AE = 0.1846 H; el tercer sitio probable es el hidroxilo unido al C7 con 1.6497 H de diferencia y el cuarto sitio de ataque es el hidroxilo unido al C5 con 3.389 H de diferencia.
En el flavonoide 2 el enlace O-H unido al C3, es el sitio más probable de ataque porque tiene el valor de entalpia menor de disociación de enlace tabla N°7 (13.6754 Hartree) por lo que se le asigna el valor de 0.0 H ver figura 7; el grupo hidroxilo unido al C5 es el segundo sitio probable con un AE = 3.7746 H.
En el flavonoide 3 el enlace O-H unido al C4, es el sitio más probable de ataque porque tiene el valor de entalpia menor de disociación de enlace tabla N°7 (10.28596 Hartree) por lo que se le asigna el valor de 0.0 H ver figura 7; el grupo hidroxilo unido al C5 es el segundo sitio probable con un AE = 5.324 H.
En el flavonoide 4 el enlace O-H unido al C6 es el sitio más probable de ataque porque tiene el valor de entalpia menor de disociación de enlace tabla 7 (11.1077 Hartree) por lo que se le asigna el valor de 0.0 H ver figura 7; el grupo hidroxilo unido al C3, es el segundo sitio probable con un AE =0.398 H; el tercer sitio probable es el hidroxilo unido al C7 con 1.504 H de diferencia, el cuarto sitio de ataque es el hidroxilo unido al C4, con 2.18 H de diferencia y el ultimo sitio de ataque es el hidroxilo unido al C5 con 3.013 H de diferencia.
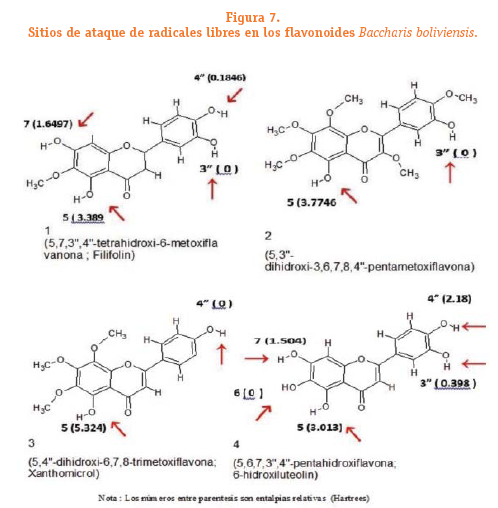
Con base a los datos anteriores podemos plantear que el flavonoide 4 tiene mayor capacidad antioxidante debido al mayor numero de grupos hidroxilo y a los valores bajos de EDE relativa respecto al flavonoide 1 que ocuparía el segundo lugar en términos de capacidad antioxidante, apoyan a estos resultados el hecho de considerar los valores de la segunda diferencia de EDE entre estos dos flavonoides se obtiene una diferencia de 1.106 H para el flavonoide 4 y 1.4651 H para el flavonoide 1. Los flavonoides 2 y 3 ocuparían las posiciones 3 y 4 en términos de capacidad antioxidante respectivamente.
Los resultados obtenidos se presentan en la Tabla 8.
Orbitales moleculares
Se realizó un cálculo de las energías de los orbitales moleculares de alta y baja energía HOMO (Highest Occupied Molecular Orbital) u Orbital Molecular de Ocupado más alto y LUMO (Lowest Unoccupied Molecular Orbital) u Orbital Molecular Vacío o Vacante más bajo. La diferencia de energías del HOMO y LUMO se denomina salto de banda, lo cual puede servir como una medida de la excitabilidad molecular. A menor energía, más fácilmente puede ser excitada.
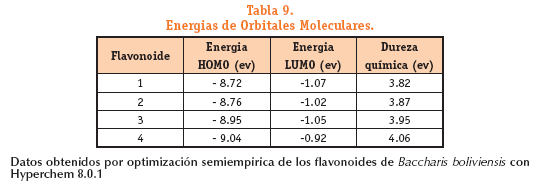
El valor de energía HOMO en los flavonoides estudiados de la tabla anterior muestra que el flavonoide 4 presenta el menor valor de energía por tanto es la molécula más fácilmente excitable.
Todas presentan un valor alto de LUMO en comparación con la energía HOMO Esto demuestra la disponibilidad molecular para la participación en interacciones electrónicas.
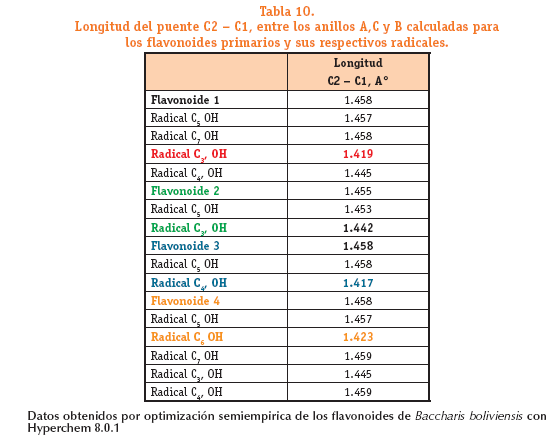
La actividad antioxidante en los flavonoides no solo está relacionada con el número de hidroxilos, factores como el grado de deslocalización electrónica (acortamiento del enlace C2 - C1, entre los anillos A,C y B influyen en esta actividad. A continuación mostramos los valores de longitud del p
Los cuatro flavonoides presentan una geometría molecular plana.Los Ángulos de torsión entre O1 - C2 - C1, - C6, para el flavonoide 1 es de 0.23°, para el flavonoide 4 es de 0.20°, para el flavonoide 2 es de 4.88° y para el flavonoide 3 de 9.62°
La actividad antioxidante en los flavonoides no solo está relacionada con el número de hidroxilos, factores como el grado de deslocalización electrónica (acortamiento del enlace C2 - C1,) entre los anillos A,C y B influyen en esta actividad.
Esta estabilidad de las estructuras planas de estos cuatro flavonoides es debido a la deslocalización electrónica que existe coadyuvado con el buen traslape de los orbitales moleculares entre los anillos A,By C.
Las cargas negativas correspondientes a los oxígenos en la estructura de los cuatro flavonoides estudiados proporcionan una primera idea del lugar donde se formara el radical más estable de cada estructura.
Las cargas negativas correspondientes a los oxígenos confirman el lugar donde se forman los radicales más estables en cada estructura. Esta predicción es correcta para todas las estructuras; radical C3, del flavonoide 1 (-0.501); C3, del radical C3, del flavonoide 2 (-0.481); C4, del radical C4, del flavonoide 3 (-0.439) y C6 del radical C6 del flavonoide 4 (-0.537)
En cuanto a la reactividad el sitio más reactivo del filifolin corresponde al grupo OH unido al C3,, para el flavonoide 2 la posición más reactiva se encuentra en el grupo OH unido al C3,; en el flavonoide 3 el sitio más reactivo es el grupo hidroxilo OH que se encuentra unido al C4, y el sitio más reactivo en el flavonoide 4 es el grupo hidroxilo unido al C6.
La reactividad de estos flavonoides no solo depende del número y posición del OH en la estructura, también depende de las diferencias en las entalpias de disociación de enlace O-H entre todos los posibles radicales generados.
En cuanto a la actividad antioxidante entre los cuatro flavonoides podemos plantear que el flavonoide 4 tiene mayor capacidad antioxidante debido al mayor número de grupos hidroxilo y a los valores bajos de EDE relativa respecto al flavonoide 1 que ocuparía el segundo lugar en términos de capacidad antioxidante, apoyan a estos resultados el hecho de considerar los valores de la segunda diferencia de EDE entre estos dos flavonoides se obtiene una diferencia de 1.106 H para el flavonoide 4 y 1.4651 H para el flavonoide 1. Los flavonoides 2 y 3 ocuparían las posiciones 3 y 4 en términos de capacidad antioxidante respectivamente.En suma la actividad antioxidante de los cuatro flavonoides estudiados presenta el siguiente orden: flavonoide 4 > flavonoide 1 > flavonoide 2 > flavonoide 3
El valor de energía HOMO en los flavonoides estudiados muestra que el flavonoide 4 presenta el menor valor de energía por tanto es la molécula más fácilmente excitable.
Todas presentan un valor alto de LUMO en comparación con la energía HOMO. Esto demuestra la disponibilidad molecular para la participación en interacciones electrónicas.
El acortamiento del enlace C2 - C1, en los radicales respecto a sus estructuras primarias, indica que existe un mayor flujo electrónico a través de este enlace desde el anillo C al B o viceversa.
REFERENCIAS BIBLIOGRÁFICAS
Almanza,G;Yupanqui,J; Calle,A; Flores, Y;(2012). Flavonoides de Baccharis boliviensis.Revista Boliviana de Quimica.Vol 29, N°2pp155-160 [ Links ]
Allen RG, tresini M.(2000) Oxidative stress and gene regulation. Free Radic Biol Med.;28:463-499. [ Links ]
Beckman K B, AMES B N.(1998) The free radical theory of aging matures. Physiol Rev. pp55-65 [ Links ]
Balsano C, Alisi A.(2009) Antioxidant effects of natural bioactive compounds. Curr. Pharm Des.2009; 15(26):3063-73. [ Links ]
Ferretti G, Bacchetti T, Belleggia A, Neri D.(2010) Antioxidants: from farm to ta-ble. Molecules.15(10): 6993-7005 [ Links ]
Finkel., T. & Holbrook, N.J.(2000) "Oxi-dants, oxidative stress and the biology of ageing", en Nature. 408, pp. 239-247. [ Links ]
Guillen F,Ruiperez I.Manual de Geriatría 3°Ed.(2002) Envejecimiento celular y molecular: Teorías del envejecimiento. Barcelona:Masson;p.29-42 [ Links ]
Lewars, E.(2011) Computational chemistry. Introduction to the theory and applica-tions of molecular and quantum mecha-nics, 2nd ed., Springer, pp.30-45 [ Links ]
Page, A. J., Ohta, Y., Irle, S., Morokuma, K.(2010) Mechanisms of single-walled carbon nanotube nucleation, growth, and healing determined using QM/MD methods. Accounts of Chemical Research 43 (10): 1375-1385. [ Links ]
Perez,G.(2003) Los flavonoides.Rev.Medica inv.cubana.22(1):48-57 [ Links ]
Miquel J. (1989) Historical introduction of free radical and antioxidants biomedical researcj. En: Miquel J, Quintanilha AT, Weber H (eds): CRC Handbook of free radicals and antioxidants. Vol 1.Boca Ratón,FL: CRC press;.p.3-13 [ Links ]
Rybczynska M. (1994) Biochemical aspects of free radical medited tissue injury. Postepy Hig Med. Dows 48:419-41. [ Links ]
Sakata, K; Saito, N; Honda, T,(2006) Tetrahe-dron.62,3721 [ Links ]
Sies H. (1986) Biochemistry of oxidative strss.Angeq Chem 25:1058-1071. [ Links ]
Sánchez R, A. y cols. (2000) Quimica Com-putacional. U.Malaga.pp 80-82 [ Links ]
Suárez, D y cols. (2000) Objetivos y características de la Quimica Computacional. Bol Grupo Español carbón. pp 23-33 [ Links ]
