










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO  uBio
uBio Compartir

 Permalink
PermalinkRevista CON-CIENCIA
versión impresa ISSN 2310-0265
Rev.Cs.Farm. y Bioq vol.1 no.1 La Paz oct. 2013
ARTÍCULOS DE INVESTIGACIONES FACULTATIVOS DOCENTE
Biodisponibilidad y bioequivalencia in vitro en cápsulas de amoxicilina de 500 mg comercializados en Bolivia
DAZA CALDERÓN, MARIA LUISA1
1 Facultad de Ciencias Farmacéuticas y Bioquímicas, Universidad Mayor de San Andrés, La Paz-Bolivia, mldazacal@hotmail.com
Resumen
Los estudios de Biodisponibilidad y Bioequivalencia permiten demostrar si el principio activo tiene el mismo desempeño farmacocinético en el medicamento genérico que en el innovador ó con el de referencia establecido en un país, para así garantizar a través de estos estudios la in-tercambiabilidad, que deben ser desarrolladas como una exigencia a los productos genéricos, como una verdadera Garantía de Calidad de los medicamentos, así lo estipula la Ley del Medicamento y las Buenas Prácticas de Manufactura aprobadas en Bolivia a partir de diciembre de 1996.
La amoxicilina, antibiótico de amplio espectro muy utilizado en nuestro país, pertenece a la clase I en el Sistema de Clasificación Biofarmacéutica y por tanto justifica la realización de estudios de Biodisponibilidad y Bioequivalencia in vitro, por medio de perfiles de disolución.
Para este estudio se eligen cápsulas de 500 mg de 4 laboratorios nacionales y extranjeros, de mayor venta en el mercado boliviano en el año 2003.
A los 4 productos (3 lotes por producto) se realiza el protocolo de control de calidad. Luego para elegir los lotes que entran al estudio de Bioequivalencia se realiza el Análisis Cinético de los perfiles de disolución para determinar la constante de velocidad de disolución intermedia de los tres lotes y elegir el lote de comportamiento intermedio para que entre al estudio de Bioequivalencia. Los perfiles de disolución de los cuatro lotes elegidos se comparan a través del método estadístico Modelo Independiente con factor de similitud. Se concluye que los 4 son bioequivalentes e intercambiables entre sí.
Palabras clave
Biodisponibilidad, Bioequivalencia, Perfiles de Disolución.
Abstract
The Biodisponibility and Bioequiva-lence studies allow to demonstrate if the active principle has the same pharmacoki-netic performance in the generic drug as the standard set up in a country, to con-sider them interchangeable and to guar-antee through these tests that they must be developed as a demand to the generic drugs. In this way to contribute to have a true Guarantee of Quality of drugs like the Drugs Law and the Good Manufacture Practices were been approved in Bolivia of approved in Bolivia on December of 1996.
The amoxycillin is an antibiotic of wide spectre very used in our country, it belongs the first class in the Biopharmaceutical Classification System (BCS), and therefore the execution of in vitro Biodisponibility and Bioequivalence studies by dissolution profiles have been justified.
An in vitro Biodisponibility and Bio-equivalence study of the amoxycillin trihy-drate (capsules of 500 mg) of four national and foreign laboratories of more sale in the Bolivian market was carried out in this re-search, through an experimental and statis-tical design, set up in official documents of the United States Pharmacopoeia (USP).
To the four products (three lots for product) were made the analysis of quality control for solid oral pharmaceutical forms, then to choose the lots that enter to bio-equivalence study it makes the Kinetic Analysis of the dissolution profiles of dissolved percentage accumulated versus time with a valuated method analytic, and these profiles are compared through the statistical method, IndependentModel with the Similarity Factor. It concludes that the products of the study are bioequivalentand interchangeable to each other, since in our country the innovative product is not marketed and it doesn't have a product of Reference settled down by the competentauthorities.
KEY WORDS
Biodisponibility, Bioequivalence, dissolution profiles.
INTRODUCCIÓN
Biodisponibilidad y Bioequivalencia, dos conceptos muy utilizados en la actualidad, están íntimamente ligados con el desarrollo de dos nuevas ciencias, la Biofarmacia y la Farmacocinética, que surgieron en la primera mitad del siglo pasado, y alcanzaron su auge en las décadas de los 60 a 80 del siglo XX.
La Biodisponibilidad, se define como la velocidad y la cantidad de un fármaco que alcanza la circulación sistémica a partir de una forma farmacéutica.
El término de Bioequivalencia se refiere a la velocidad y proporción en que el mismo principio activo, proveniente de dos medicamentos "iguales" alcanza la circulación sistémica; por ende, un estudio de Bioequivalencia es un estudio de Biodisponibilidad relativa, en el cual se compara la velocidad y la cantidad absorbida de productos que contienen el mismo fármaco (misma sal), la misma dosis, en la misma forma farmacéutica (Informe, 1994).
Para ello, se deberá demostrar, que no existen diferencias estadísticamente significativas en los productos provenientes de diferentes marcas, ó productos genéricos, frente al producto innovador ó el producto de referencia.
El medicamento original o innovador es aquél que contiene un principio activo nuevo y con el que se ha realizado una investigación y desarrollo completo, desde su síntesis química hasta su utilización clínica, es por tanto, el primero, y a veces el único, que aporta datos propios de seguridad y eficacia terapéutica del principio activo, administrado en una especialidad farmacéutica concreta, a dosis determinada y con indicaciones específicas.
El producto de referencia es un producto farmacéutico en relación con el cual el nuevo producto pretende ser intercambiable, se puede usar el producto que sea líder el mercado y haya recibido autorización para comercializarse y estén documentados su eficacia, inocuidad y calidad (Center for Drug Evaluation and Research CDER.FDA, 1997)
A nivel mundial se inicia un proceso de armonización con el propósito de reducir requisitos innecesarios y duplicados para la aprobación de fármacos para la venta, y para acelerar la disponibilidad y reducir el costo de los fármacos (Informe, 1994).
Se instruye a la Agencia Europea para la Evaluación de Productos Medicinales (EMEA), como el organismo para regular, coordinar el proceso de armonización con respecto a los requisitos de aprobación de productos medicinales y al Comité Internacional de Armonización (ICH), conformado entre la Comunidad Europea, Japón y Estados Unidos y la Organización Panamericana de la Salud (PAHO), para promover la organización en las Américas, a través de la Red Panamericana de las Agencias Regulatorias de Fármacos, para el Proceso de Armonización Regulatoria (PANDRA), con el objeto de coordinar el proceso de armonización en las Américas, conformado por un grupo de trabajo que se encarga de temas relacionados a la Biodisponibilidad y Bioequivalencia (BA/BE) (FDA, 2001).
El Comité de Expertos de la Organización Mundial de la Salud (OMS), en Especificaciones para las Preparaciones Farmacéuticas, se reunió en Ginebra el 28 de septiembre de 1994, para abordar la amplia variedad de asuntos relativos a la garantía global de la calidad de los Productos Farmacéuticos. En el Anexo 9 del Informe 34, se dan las directrices sobre los requisitos de registro para establecer el carácter intercambiable del genérico frente al innovador o de referencia (1). Posteriormente (2006) en el Anexo 7 del Informe 40 presenta la Guía de los Requerimientos de Registro para establecer Intercambia-bilidad. y la propuesta para obviar los requerimientos de Bioequivalencia in vivo para formas de dosificación oral de la lista modelo de los medicamentos esenciales de liberación inmediata (Informe 40, 2006).
Los productos genéricos deben satisfacer las mismas normas de calidad, eficacia e inocuidad que el producto de origen. Además, se debe proporcionar una garantía razonable de que son clínicamente intercambiables con productos existentes en el mercado y que son nominalmente equivalentes. El carácter intercambiable, se garantiza suficientemente aplicando las Buenas Prácticas de Manufactura y comprobando la observancia de las especificaciones pertinentes de la farmacopea. Todas las formas farmacéuticas orales sólidas deben demostrar equivalencia terapéutica y los datos correspondientes se incluirán en la documentación presentada junto con la solicitud de autorización de comercialización (Informe, 1994).
Los organismos de reglamentación deben exigir la documentación que acredite que un producto farmacéutico genérico satisface los siguientes tres conjuntos de criterios:
Fabricación según Buenas Prácticas de Manufactura (BPM) y Control de Calidad
Características y etiquetado del producto
Equivalencia terapéutica
La evaluación de la Bioequivalencia normalmente exigirá un estudio in vivo, o una justificación de que dicho estudio no se requiere en un caso particular. Entre los tipos de estudios in vivo pueden mencionarse los estudios de Bioequivalencia, los estudios farmacodinámicos y los ensayos clínicos comparativos. En ciertos casos, los estudios in vitro pueden bastar para aportar equivalencia terapéutica (Informe, 1994).
La amoxicilina antibiótico de amplio espectro actúa bloqueando la actividad transpeptidasa de las Proteínas Fijadoras de Penicilina en las bacterias en fase de crecimiento, así, la síntesis de peptidoglucano (esencial en la pared bacteriana) disminuye y la bacteria muere por efecto osmótico o digerida por enzimas autolíticas (Martindale, 2005).
La amoxicilina trihidrato en cápsulas de 500 mg, producto genérico y esencial, tanto de procedencia nacional como de importación comercializado en Bolivia, debe cumplir los mismos criterios de calidad que el innovador, porque, al carecer de ensayos clínicos propios, basan sus datos de seguridad y eficacia terapéutica presentada en la documentación publicada que existe sobre dicho principio activo del producto innovador.
En Bolivia, hace 25 años se inicia la comercialización de este producto, la lista oficial de medicamentos con registro sanitario vigente, (año 2003), registra que se comercializa por 37 laboratorios, tanto producto de marca como genérico de procedencia nacional y de importación. (Lista Nacional de Medicamentos con Registro Sanitario Vigente, 2004).
Como producto genérico existen 15 líneas de producción, 8 de laboratorios nacionales y 7 de importación, cuya calidad Biofarmacéutica no ha sido avalada a través de pruebas de Bioequivalencia, requisito no exigido en los formularios para el registro sanitario.
En la Clasificación Biofarmacéutica, Clase I están los fármacos que son: altamente solubles y altamente permeables. Los estudios de bioequivalencia in vitro, para ciertos medicamentos y formas farmacéuticas, pueden evaluarse mediante pruebas de disolución, a través de la comparación de perfiles de disolución, como la amoxicilina en cápsulas de 500 mg (Informe 40, 2006).
PARTE EXPERIMENTAL
Metodología
Se selecciona los 4 productos de mayor venta (IMS)
Protocolo de control de calidad USP 26
Validación del método analítico: especificidad, exactitud, precisión y linealidad
Análisis estadístico ANOVA para verificación del comportamiento entre lotes
Perfiles de disolución, con siete puntos de muestreo
Análisis cinético y estadístico para determinar K (constante de disolución)
Elección del lote de K intermedia, de cada producto que entrará al estudio de BE
Diseño experimental para el estudio de Bioequivalencia con los 4 lotes elegidos.
Comparación de los perfiles de disolución por el método estadístico Modelo Independiente con factor de similitud.
RESULTADOS Y DISCUSIÓN
Resultados
Determinar los productos de mayor consumo de cápsulas de Amoxicilina trihidrato de 500 mg, tanto de procedencia nacional como de importación en Bolivia, con base en el reporte del Internacional Marketing Stadistic (IMS) de los 4 productos de mayor venta en el año 2003.
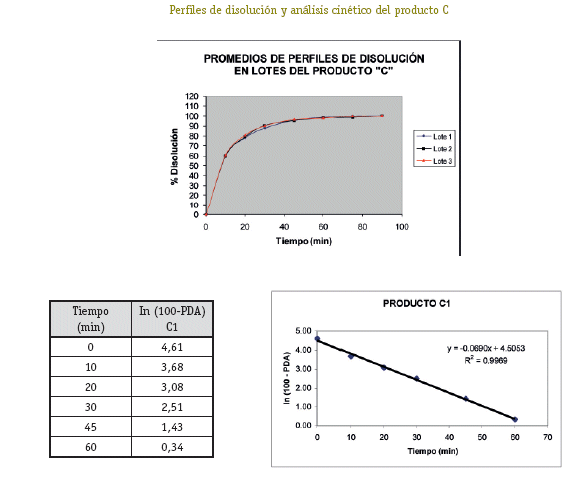
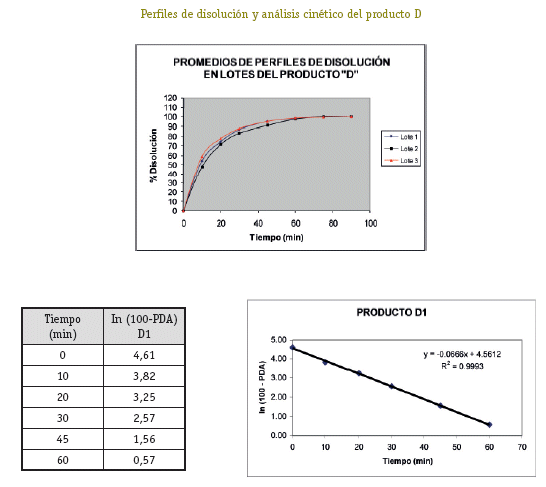
De cada uno de los productos se han seleccionado tres lotes (A1, A2, A3; B1, B2, B3; C1, C2, C3; D1, D2, D3) a los cuales se ha realizado el control de calidad para formas farmacéuticas sólidas orales tales como: Verificación de la rotulación, Prueba de hermeticidad del envase, Características organolépticas, Pruebas de Identificación, Cuantificación del principio activo, Uniformidad de contenido, Determinación de humedad y Prueba de disolución (USP 26, 2003).
Los resultados obtenidos de todos los lotes de los 4 productos cumplen con los requerimientos de calidad especificados en la monografía correspondiente a cápsulas de amoxicilina de la farmacopea USP 26.
Posteriormente se valida el método analítico, para efectuar los perfiles de Disolución de las cápsulas de Amoxicilina de 500 mg, teniendo en cuenta los parámetros de especificidad, exactitud, precisión y linealidad, para el estándar y cada uno de los lotes (Rojas, 2003 y AEFI, 2001).
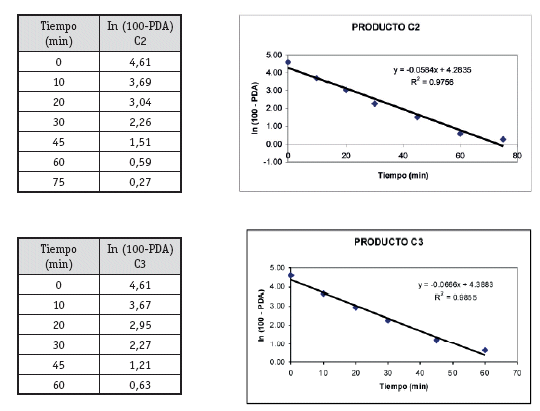
Una vez analizados los resultados de las pruebas de Control de Calidad de los 4 productos con resultados que se ajustan a los requerimientos de la farmacopea, se define pasar a la etapa de selección de los lotes que entran al estudio de Bioequivalencia in vitro, (un lote de cada producto). Estos lotes se seleccionaron primero sobre la base del análisis estadístico tipo ANOVA (método FISHER) para determinar el comportamiento de los 3 lotes de cada producto, no detectándose diferencias significativas entre los tres lotes, luego de este análisis se procedió al análisis cinético para determinar las constantes de disolución (K de disolución min-1), para elegir el lote de cada producto que tenga un comportamiento intermedio, resultando A2, B1, C1 y D1 los lotes elegidos para al estudio de Bioequivalencia in vitro.
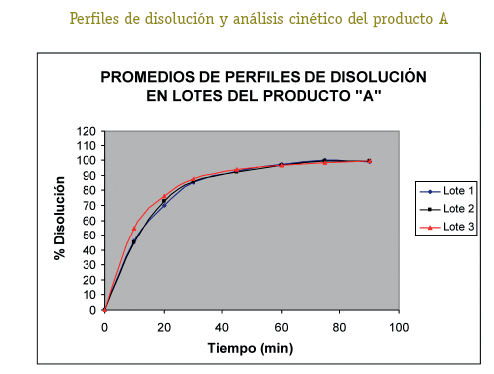
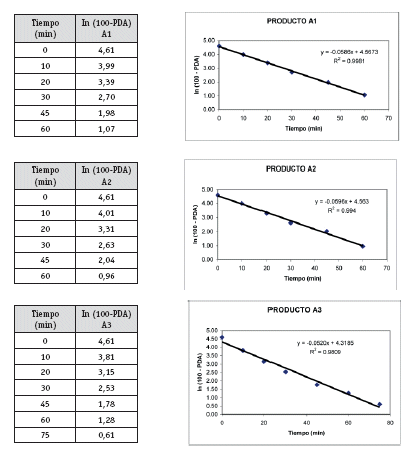
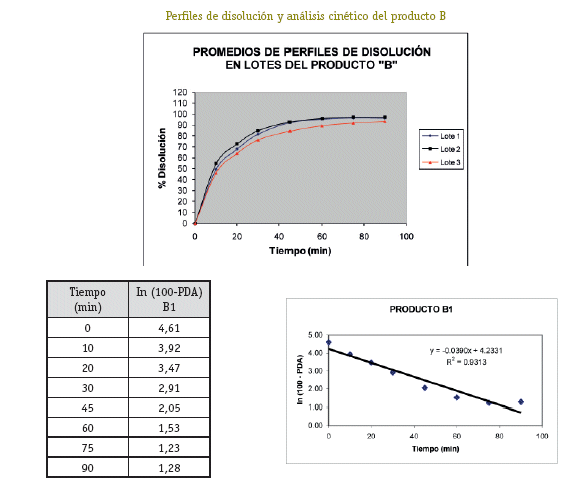
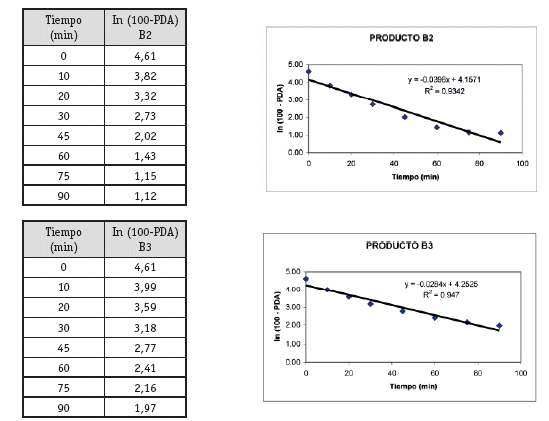
La detección de la Bioequivalencia in vitro entre los 4 productos se realizó a través del análisis estadístico mediante el Modelo Independiente calculando las ecuaciones para el factor de diferencia f1 y el factor de similitud f2 (5), siendo los valores límite del f1 (0-15) y f2 (50-100). Los resultados obtenidos de los cálculos de los factores de diferencia (f1) y similitud (f2) nos permiten afirmar que los productos A, B, C y D, se pueden considerar bioequivalentes e intercambiables entre sí, por los resultados obtenidos en los perfiles de disolución. (Cárcamo, 1981).
CONCLUSIONES
En el presente trabajo se demuestra la Bioequivalencia in vitro entre los cuatro productos seleccionados para este estudio, bajo las mismas condiciones de disolución y sobre la base de un protocolo realizado a partir de documentos internacionales que dan pautas sobre la realización de estos estudios.
Al hacer las comparaciones múltiples entre los diversos productos del estudio, a través del Modelo Independiente se demuestra que:
Los productos A, B, C y D, se pueden considerar bioequivalentes e intercambiables, esta afirmación, se basa en la comparación de los perfiles de disolución y el correspondiente cálculo de los factores de diferencia (f1) y similitud (f2) del Modelo Independiente, valores de f1 entre 0 -15 y valores de f2 entre 50 -100.
En los ensayos de control de calidad el producto B, si bien cumple con las especificaciones, la variabilidad de los resultados representada por el CV muestra valores ligeramente por encima de lo permitido en la prueba de variación de peso y uniformidad de contenido, cuyos resultados de CV están: para el lote B1 5,85%, para el lote B2 6.14%, y para el lote B3 6.96%.
El producto C, es el que presenta menor variabilidad en los resultados, tanto para control de calidad como en el estudio de Bioequivalencia in vitro, por lo que se podría proponer como Producto de Referencia.
El sistema empleado para la selección de los productos que entraron al estudio se puede considerar válido porque involucra diversos criterios que permiten considerar aquellos productos que tiene mayor uso en la comunidad en general y por lo tanto un importante riesgo para la salud en caso de no cumplir con los parámetros de calidad.
El protocolo desarrollado y el diseño experimental y estadístico utilizado para la selección de los lotes que entran al estudio de Bioequiva-lencia in vitro proporciona un método sencillo para identificar el lote de comportamiento intermedio de cada producto.
La metodología y el tratamiento estadístico presentado en esta investigación ofrecen una herramienta muy útil en el desarrollo de estudios de Bioequivalencia in vitro de medicamentos genéricos fabricados en el país ó de importación, los cuales deben cumplir con esta exigencia, para garantizar la calidad de los medicamentos que llegan a la población y su posible intercambiabilidad.
Por su facilidad en el desarrollo y confiabilidad, se propone la aceptación del procedimiento establecido en el presente trabajo de investigación para evaluar la Bioequivalencia in vitro de medicamentos que de acuerdo a la clasificación biofarmacéutica sean aceptados para este tipo de ensayos.
REFERENCIAS
INFORME 34a (1994) Comité de expertos de la OMS en especificaciones para las preparaciones farmacéuticas. Ginebra: OMS. [ Links ]
CENTER FOR DRUG EVALUATION AND RESEARCH CDER. FDA (1997) Guidan-ce for Industry. Dissolution Testing of immediate Release Solid Oral Dosage Form. Rockville, Disponible en: http://www.fda.gov/cder/guidance.htm. USA. [ Links ]
FDA. (2001) Curso regional de biodisponi-bilidad y bioequivalencia Módulos 1 y 2 Caracas [ Links ]
INFORME 40 (2006) Comité de expertos de la OMS en especificaciones para las preparaciones farmacéuticas. Ginebra: OMS. [ Links ]
MARTINDALE. (2005). Guía Completa de Consulta Farmacoterapéutica. Barcelona: Pharma Editors. Pp: 217-391 [ Links ]
LISTA NACIONAL DE MEDICAMENTOS CON REGISTRO SANITARIO VIGENTE. (2004) Dirección de Medicamentos y Tecnología en Salud. [ Links ]
FARMACOPEA DE LOS ESTADOS UNIDOS DE AMÉRICA USP 26 (2003) USA. [ Links ]
J. ROJAS. (2003) Dossier del Curso Taller VALIDACIÓN DE METODOLOGÍAS ANALÍTICAS La Paz: UMSA [ Links ]
ASOCIACIÓN ESPAÑOLA DE FARMACÉUTICOS DE LA INDUSTRIA (AEFI). (2001). Validación de Métodos Analíticos. AEFI: Barcelona pp. 39-134. [ Links ]
EDISON CID CÁRCAMO. (1981) Cinética de Disolución de medicamentos. Secretaría General de la Organización de los Estados Americanos Washington,D.C. [ Links ]
