











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Si se considera desde el tiempo de definición a la fecha, la mucopolisacaridosis (MPS) es una patología nueva, los primeros casos publicados fueron en 1917 y en un inicio se presentaba como un grupo de pacientes con alteraciones morfológicas (talla baja, hernia inguinal, facie tosca, respiración ruidosa), es a partir de 1940 que se va identificando su génesis y gracias a ello hoy en día se afirma que la Mucopolisacaridosis (MPS) es una enfermedad genética rara, con una incidencia del 0,1% dentro del grupo de enfermedades genéticas, siendo de tipo autosómico recesivo, que produce un defecto en las enzimas encargadas de la degradación de los glicosaminoglicanos, los cuales se almacenan en los lisosomas produciendo disfunción de tejidos y síntomas multiorgánicos. Se clasifica en 7 tipos que se diferencian bioquímicamente por su deficiencia enzimática asociada: MPS I, MPS II, MPS III, MPS IV, MPS VI, MPS VII, MPS IX1,2,3.
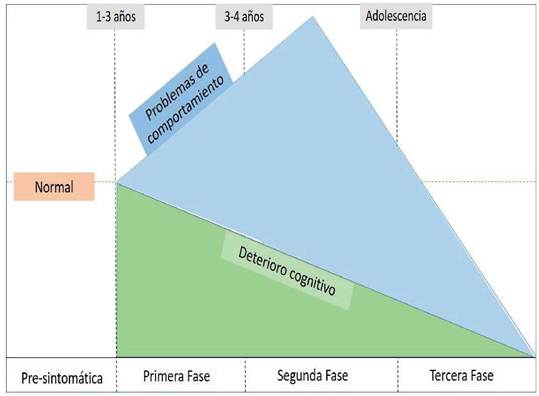
Fuente. Elaboración propia en base a la información obtenida de: Zhou J, Lin J, Leung WT, Wang L. A basic understanding of mucopolysaccharidosis: Incidence, clinical features, diagnosis, and management. Intractable Rare Dis Res2020; 9(1): 1-9. y Heon-Roberts R, Nguyen ALA, Pshezhetsky AV Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. J Clin Med 2020; 9(2): 344.
Figura 1 Historia Natural de la Enfermedad MPSIII
La importancia de los glicosaminoglicanos radica en que son componentes de la matriz y membrana extracelular, participando de igual manera en la hidratación y mantenimiento trastorno autista se ha visto relacionado con el gen OXTR (codifica el receptor de oxitocina) y el gen HRH1 (codifica receptor de histamina); la demencia y deterioro cognitivo con la disminución del receptor de insulina y factor de crecimiento insulínico tipo 1; el déficit auditivo con la proteína Homer 2 quien actúa dentro de la señalización de calcio celular11.
Clínica
Los síntomas generalmente comienzan entre los 2 y 6 años e incluyen la desaceleración en los hitos del desarrollo y la adquisición del lenguaje, con alteraciones del comportamiento que cursa con periodos de atención cortos, sin respuesta a instrucciones, ausencia de sensación de peligro y agresividad junto con alteraciones del sueño volviéndose activos las 24 horas del día12. Los síntomas pueden enmascararse como trastornos del comportamiento incluido el autismo, el trastorno por déficit de atención e hiperactividad, que ocasionan un diagnóstico incompleto o tardío9.
Al examen físico se puede apreciar una facie gruesa, hirsutismo, alteración esquelética con rigidez articular, presencia de hernia umbilical y/o inguinal, así como hepato y/o esplenomegalia; cursa con infecciones respiratorias altas, sordera y diarrea episódica5,10
Inicia con un ritmo de crecimiento normal o acelerado en el primer año, el cual va deteniéndose, evidenciando talla baja a los cinco años de edad5,13.
Respecto a los subtipos se encuentran peculiaridades, como ser MPS IIIA con mayor deterioro ocular (atrofia nerviosa, lesión corneal, retinopatía y glaucoma)8 y MPS IIIB afectación cerebral (atrofia cortical, del cuerpo calloso, sustancia blanca y cerebelo con disminución de células de Purkinje)14.
Así como puede cursar con una clínica tan compleja, puede haber casos donde cursa únicamente con distrofia retiniana de inicio tardío junto a compromiso cardíaco leve y hepatoesplenomegalia5.
Complicaciones
Tiene un impacto global, con deterioro profundo del sistema nervioso central, el sistema respiratorio, predispuesto a cuadros constantes de infecciones, alteración del sistema musculoesquelético, llevando a la rigidez y degeneración articular; y el sistema cardiaco, con afectación de las válvulas mitral y aórtica12.
Diagnóstico
La sospecha guía al diagnóstico de MPS III y el mismo se confirma mediante examen de laboratorio el cuál inicia con la determinación de glicosaminoglicanos urinarios, cuyo resultado positivo sugiere MPS, aunque un resultado negativo no lo descarta. El diagnóstico definitivo se realiza por medio del test genético molecular que combina las técnicas del panel multigene (genes específicos: GNS, HGSNAT, NAGLU, SGSH) y pruebas genómicas completas (comprehensive genomic testing)10.
Otra prueba, para aumentar la precisión diagnóstica del estudio genético, es el ensayo enzimático en leucocitos de sangre periférica y cultivo de fibroblastos cutáneos que mide la actividad de las enzimas GNS, HGSNAT, NAGLU y SGSH10. Dicha prueba puede aplicarse en muestras de vellosidades coriónicas o amniocentesis para diagnóstico prenatal, también en muestra de sangre fetal seca para tamizaje en recién nacidos15 (verfigura 2).
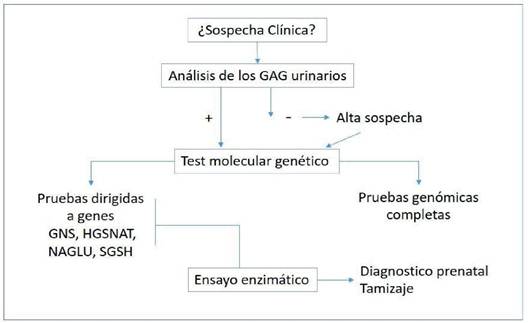
Fuente. Elaboración propia en base a información obtenida de Wagner VF, Northrup H. Mucopolysaccharidosis Type III.2019. En: Adam MP, Ardinger HH, Pagon RA, et al. GeneReviews® Seattle (WA): University of Washington, Seattle; 1993-2020 y Jones S, Wynn R. Mucopolysaccharidoses: Clinical features and diagnosis. UpToDate2019
Abreviaturas: GAG: Glicosaminoglicanos; GNS: N-acetilglucosamina 6-sulfatasa; HGSNA: a-glucosaminidasa acetiltransferasa; NAGLU: a-N-acetilglucosaminidasa; SGHS: heparan-N-sulfatasa.
Figura 2 Métodos diagnósticos MPS III
Los estudios de imagen son variables, se puede encontrar: en la radiografía disostosis múltiple, deformidad de cadera; en la resonancia magnética alteración de la sustancia blanca, atrofia cerebral difusa; y en el ecocardiograma alteración de la válvula mitral o aórtica10.
Tener presente que el diagnóstico puede llegar a retrasarse alrededor de 2 años desde el inicio de los síntomas por confundirlas con otras patologías, como lo refleja un estudio realizado en México que reportó que los casos de MPS cursaron con su primera manifestación clínica alrededor de los 3 años de edad y fueron diagnosticados alrededor de los 5 años16, y que el diagnóstico temprano en la población de progreso lento es más difícil, pudiendo permanecer sin diagnóstico hasta la edad adulta9.
Tratamiento
Actualmente no se cuenta con un tratamiento curativo, se enfoca en brindar medidas paliativas y de soporte, con manejo multidisciplinario de diversas especialidades, se debe implementar terapias motoras, conductales, así como uso de medicación específica para la diversa sintomatología que presenta, como ser: carbamazepina para convulsiones, neurolépticos en alteración del comportamiento y trastornos del sueño, antibióticos para los cuadros de infecciones frecuentes; puede llegar a requerir intervención quirúrgica, como ser una gastrostomía por cuadro de desnutrición o descompresión medular, debido a disostosis toracolumbar7,9.
Los pacientes deben de contar con vigilancia constante, junto a un ambiente seguro para evitar que se lesione, su esperanza de vida oscila entre los 20 a 30 años, aunque hay reportes de hasta los 70 años y su principal causa de mortalidad son las infecciones del tracto respiratorio (neumonía) y la afectación neurológica9,10.
Es prudente que la familia cuente con grupos de apoyo, con evaluaciones frecuentes de su salud mental, ya que el cuidado de los pacientes desencadenaba cuadros de agotamiento mental, depresión, ansiedad y estres9.
Si bien no se cuenta con un tratamiento curativo, se están desarrollando terapias en busca de disminuir el deterioro del paciente, mencionamos el estudio comparativo en pacientes con MPS IIIA realizado en el Centro Internacional de Trastornos Lisosomales (ICLD) del Centro Médico Universitario de Hamburgo- Eppendorf Alemania, que determino que el trasplante de células madre hematopoyéticas en edades tempranas limito la disminución de la función motora y cognitiva17. Otros reportes indican que el uso de trasplante de células madre hematopoyéticas antes del inicio de la enfermedad son útiles para tratar los síntomas somáticos, pero no son efectivas para prevenir la neurodegeneración en pacientes. Por otro lado, dos ensayos clínicos basados en la inyección intracerebral de virus adenoasociados mostraron algunas mejoras neurológicas en pacientes de los subtipos IIIA y IIIB2.
Un elemento que puede llegar a influir en la respuesta a los diversos ensayos terapéuticos que se están realizando es el fenotipo, porque si es de progresión lenta cursa con síntomas leves y vida útil más larga a diferencia de los de progresión rápida9. Por tanto, si bien las nuevas investigaciones son prometedoras, no brindan un resultado concluyente.
DISCUSIÓN
Los trastornos metabólicos se han constituido como la génesis de las enfermedades genéticas, en el caso de la MPS III, se produce trastornos por almacenamiento lisosomico que afectan la degradación de los glicosaminoglicanos, y dependiendo del glicosaminoglicano afectado se clasifican en los siete diferentes grupos, incluso, actualmente se está denominando síndrome de mucopolisacaridosis plus al cuadro de síntomas típicos de MPS convencional que desarrollan otras características como defectos cardíacos congénitos, trastornos renales y hematopoyéticos18.
La base de su presentación clínica se debe a la acumulación del HS y los efectos que llega a producir, por ejemplo, la afectación neurológica se atribuye a la acumulación de HS en el cerebro que produce neuroinflamación, alteración de la señalización neuronal y muerte celular, lo que da como resultado el deterioro cognitivo, alteración del comportamiento y ataques epilepticos19; otro ejemplo es a nivel del metabolismo óseo, la acumulación de HS produce inflamación y un anormal remodelado óseo, teniendo como resultado escoliosis, fracturas patológicas, rigidez articular, síndrome del túnel carpiano y necrosis avascular de la cabeza femoral20,21.
Se señala a las infecciones como principal causa de defunción, un estudio realizado desde 1957 al 2020 en Inglaterra sobre las causas más frecuentes de defunción en pacientes con MPS III reporto que el 50,7% se debían a causas pulmonares, 5,9% a complicaciones gastrointestinales, 4,5% por insuficiencia cardíaca/congestiva, 4,5% por epilepsia/convulsiones y 2,3% por paro cardíaco22.
Si bien aún no se cuenta con un tratamiento curativo, se desarrollan terapias experimentales como el trasplante de células madres hematopoyéticas o reemplazo enzimático con la finalidad de disminuir el avance de la patología23.
Se recomienda realizar estudios de cribado tanto a los padres como a los familiares de primer y segundo grado de pacientes con MPS III para lograr una correcta asesoría genética, así como planificación familiar en pro de aumentar su autonomía reproductiva24.
CONCLUSIONES
Se define a MPS III como una enfermedad rara, la más frecuente entre todas las MPS, caracterizada principalmente por la degeneración progresiva del SNC; las características clínicas que orientan a una sospecha diagnóstica son: retraso en los hitos del desarrollo, alteración del comportamiento, infecciones frecuentes o diarreas recurrentes; tener presente que dentro de sus diagnósticos diferenciales se encuentran el trastorno de déficit de atención, hiperactividad o autismo; por lo cual, es imprescindible realizar una correcta historia clínica, profundizando en los antecedentes familiares de ambos padres. Dentro de los estudios de laboratorio se debe de realizar pruebas de glicosaminoglicanos urinarios, y se confirma por test molecular genético que combina pruebas dirigidas a genes (panel multigene) y pruebas genómicas completas; acotando que el ensayo enzimático es una herramienta que ayuda a identificar el tipo de glicosaminoglicano faltante, siendo útil también para el diagnóstico prenatal y testeo al nacimiento. Si bien no existe un tratamiento que detenga el curso de la enfermedad, lo que se busca es otorgar medidas paliativas, a través de un manejo multidisciplinario, para mejorar la calidad de vida del paciente y la familia.

