











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La agenesia pulmonar es una anomalía pulmonar poco frecuente que suele detectarse en la infancia1; posee una incidencia de un caso por cada 10 000 a 15 000 nacimientos. Se clasifica en: agenesia, aplasia e hipoplasia, formando parte de las anomalías del desarrollo pulmonar, lo cual se considera un hallazgo infrecuente en la edad adulta.2,3
Según varios autores, la prevalencia del género y la etiología difieren; la agenesia pulmonar bilateral llega a ser incompatible con la vida.4 En el caso que sea unilateral, puede ser asintomática o sintomática. Su pronóstico va a depender en gran medida si está asociada con otras malformaciones, lo cual puede empeorar presentando un cuadro clínico que puede ir desde la dificultad respiratoria aguda hasta la muerte. Algunos pacientes pueden presentar infecciones pulmonares recurrentes o puede ser un hallazgo incidental; dependiendo de las malformaciones congénitas asociadas y la buena higiene pulmonar5.
PRESENTACIÓN DEL CASO
Paciente femenina de 40 años, ama de casa, con antecedentes de asma bronquial hace 33 años, con una hospitalización previa a los 23 años por crisis asmática, diabetes mellitus tipo 2 diagnosticada hace 5 años, tuberculosis pulmonar (TBC) y sin datos de hábito tabáquico. Atendida en nuestro Hospital del Tórax por presentar dolor torácico, disnea de medianos esfuerzos y fiebre de un mes de evolución.
A su llegada al Hospital la exploración física reporto una presión arterial alterada, tórax asimétrico, disminución de la expansibilidad torácica bilateral a la palpación, matidez en hemitórax izquierdo y resonancia en derecho, a la auscultación del tórax se encuentra soplo tubárico en hemitórax izquierdo, ritmo de galope y disminución del murmullo vesicular.
El hemograma de ingreso a la institución se encuentra dentro de límites normales al igual que la química sanguínea, con excepción de la glucosa (hiperglucemia: 237 mg/dL).
En la radiografía de tórax fue encontrada opacidad homogénea que ocupa la mitad del campo pulmonar izquierdo, que borra el ángulo costofrénico y tambien se observó signos de hiperinsuflación del campo pulmonar derecho (Figura 1).
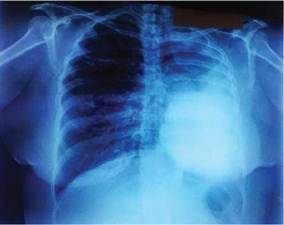
Figura 1: Radiografía PA de tórax: opacidad homogénea en campo pulmonar medio e inferior izquierdo con borramiento del surco costofrénico asociado a desplazamiento de las estructuras del mediastino a la izquierda y signos de hiperinsuflación del pulmón derecho.
La paciente es valorada por el servicio de neumología siendo ingresada bajo el diagnóstico de atelectasia pulmonar izquierda y reactivación de tuberculosis en estudio, esto en vista de los hallazgos clínicos y radiológicos iniciales.
El cultivo aerobio de micobacterias y citológico del esputo fue negativo, al igual que la GeneXepert.
La tomografía computarizada de tórax (TC) sin contraste confirma en pulmón derecho: hipertrofia importante del campo derecho inclusive se extiende hasta el hemitórax izquierdo con presencia de múltiples granulomas calcificados; pulmón izquierdo: apenas perceptible el bronquio principal izquierdo (Figura 2). Estos hallazgos producen importante desplazamiento de las estructuras del mediastino hacia el lado izquierdo, junto con estructuras vasculares: aorta torácica desviada de forma significativa hacia el lado izquierdo (Figura 2).
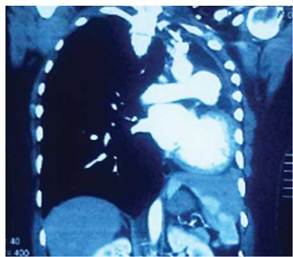
Figura 2: Tomografía computarizada de tórax: herniación del parénquima pulmonar derecho hacia el hemi tórax izquierdo.
Se indica broncoscopia donde se confirma árbol bronquial derecho normal y árbol bronquial izquierdo con amputación en entrada de bronquio.
En el electrocardiograma (ECG) se evidencian hallazgos de taquicardia sinusal, desviación a la derecha, Infarto antiguo en miocardio lateral alto, motivo por el cual se solicitó el ecocardiograma para descartar otras malformaciones congénitas o enfermedades adquiridas y se mostró miocardiopatía dilatada con disfunción sistólica grado III e hipertensión pulmonar secundaria. La prueba de función pulmonar muestra un patrón obstructivo con ligera reversibilidad con broncodilatadores.
El diagnóstico de la agenesia pulmonar izquierda es realizado durante la hospitalización, en vista de presentar proceso bacteriano se manejó con Ceftazidime por 10 días y manejo de soporte por broncoespasmo; es dada de alta a los 14 días de hospitalización por presentar mejoría clínica.
DISCUSIÓN
La agenesia pulmonar fue descrita por primera vez en 1673, siendo una malformación congénita rara con una incidencia de 1 por cada 10 000 a 15 000 autopsias, sin predominio de sexo; suele ser unilateral y es más frecuente la agenesia pulmonar izquierda; sin embargo, algunos autores describen que puede ocurrir en uno u otro lado3.
Según Boyden, la agenesia pulmonar se clasifica en 3 tipos: Tipo 1 o Agenesia: consiste en la ausencia total de parénquima, bronquio y arteria pulmonar; Tipo 2 o Aplasia: ausencia de parénquima pulmonar con presencia de bronquios rudimentarios y el Tipo 3 o Hipoplasia: presencia variable y poco desarrollada de parénquima pulmonar, árbol bronquial y vascular 1,6.
Se desconoce su etiología, sin embargo, se han propuesto diversos factores como ser: factores genéticos (trisomía parcial de los cromosomas 2p y 21q), factores teratogénicos, infecciones virales y factores mecánicos.3
También se ha descrito que, durante la embriogénesis, en la 4 semana de gestación hay un flujo sanguíneo anormal en el arco aórtico que condiciona a esta patología7; De igual manera se asocian factores de riesgo durante el embarazo como: la deficiencia de vitamina A, ácido fólico, empleo de salicilatos o con el tabaquismo3,8. Asimismo se ha detallado que durante el estadio canalicular temprano del desarrollo pulmonar, el oligohidramnios puede causar defectos.3
La agenesia bilateral implica una falla temprana en el proceso de morfogénesis bronquial y pareciera que el factor de crecimiento de fibroblastos 10 tuviera un importante papel en este estadio, al igual que la deficiencia en su receptor 2 de factor de crecimiento de fibroblastos (FGFR2); claramente la agenesia bilateral es fatal3. Esta condición usualmente se diagnostica en la niñez, en vista de que el paciente padece infecciones respiratorias a repetición, aunque una persona puede llegar a la edad adulta sin tener un diagnóstico y con una vida normal si se mantiene asintomática1.
Al nacimiento en algunos casos la agenesia se manifiesta como síndrome de insuficiencia respiratoria aguda, en otros el paciente se mantiene asintomático hasta la edad adulta, cuando se detecta incidentalmente la malformación en algún examen de rutina3,6.
Se sospecha el diagnóstico por la ausencia o disminución de los ruidos respiratorios en uno de los hemitórax; en la radiografía de tórax se muestra opacidad total en un hemitórax, hiperinsuflación compensada y herniación del pulmón contralateral, hiperlucidez retroesternal y desplazamiento posterior de las estructuras mediastinales. El diagnóstico se confirma con tomografía computarizada e idealmente con angiotomografía o angiografía5.
La realización de un ecocardiograma y ecografía abdominal es fundamental para descartar otras malformaciones, como el riñón en herradura, persistencia del conducto arterioso y el síndrome VACTERL, síndrome caracterizado por la presencia de malformaciones congénitas como anomalías vertebrales, atresia anal, defectos cardiovasculares, malformaciones traqueo-esofágicas, malformaciones renales y polidactilia; generalmente este síndrome se relaciona con la agenesia de pulmón derecho1. El diagnóstico se puede realizar mediante ecografía obstétrica morfológica en la atención prenatal8.
La fibrobroncoscopía ayuda a confirmar la terminación en fondo de saco de los bronquios afectados; las pruebas de función pulmonar suelen ser normales1,6.
Entre los diagnósticos diferenciales se deben considerar: Tetralogía de Fallot, estenosis arteria pulmonar, atelectasia pulmonar, eventración diafragmática, hernia diafragmática, neumonía masiva, situación tardía postneumonectomía, hipoplasia pulmonar, malformación adenoidea quística, fibrotórax pleurógeno, fibrotórax tuberculoso, secuestro pulmonar y Síndrome Swyer- James MaGLeod3,9. En el caso presentado, el diagnóstico diferencial se hizo con atelectasia de pulmón izquierdo.
En caso de que el paciente se encuentre asintomático, no es necesario administrar tratamiento médico, La principal complicación suelen ser las infecciones respiratorias, en vista de que no se produce un barrido adecuado de las secreciones bronquiales, estas deben ser tratadas precoz y efectivamente con antibióticos1,10.
Siempre y cuando el paciente mantenga una buena higiene pulmonar y las infecciones de la vía respiratoria sean manejadas correctamente de forma precoz, puede tener una vida completamente normal1.
Si el paciente presenta descompensación cardiovascular severa o recurrente, se puede emplear con éxito la implantación de un expansor tisular en el hemitórax afectado para evitar el desplazamiento del mediastino, lo que en teoría también permitiría una asistencia ventilatoria más agresiva (en caso de ser necesario), sin riesgo de comprometer severamente el gasto cardiaco7.
La hipertensión pulmonar es una complicación que requiere atención debido a una reducción en el lecho vascular pulmonar, que si se asocia con una cardiopatia congénita (derivación izquierda-derecha) puede progresar a una enfermedad vascular irreversible, por lo tanto, es una complicación que requiere atención6.
En general el pronóstico no es favorable, se estima un 33% de mortalidad durante el primer año de vida y el 50% en los primeros 5 años de vida. Las principales causas de mortalidad son infecciones graves del único pulmón y malformaciones cardiacas y/o de grandes vasos3. En el presente caso la ausencia de otras malformaciones asociadas y de lateralidad izquierda en nuestra paciente la sobrevida es de 40 años.

