










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Científica Ciencia Médica
versión impresa ISSN 1817-7433versión On-line ISSN 2220-2234
Rev Cient Cienc Méd v.23 n.2 Cochabamba 2020
CASO CLÍNICO
REPORTE DE CASOS DE ICTIOSIS LAMINAR EN HONDURAS; DESAFÍO DIAGNÓSTICO.
CASES REPORT OF LAMELLAR ICHTHYOSIS IN HONDURAS; CHALLENGE DIAGNOSIS.
Steven Josue Navarro Turcios1, Suyapa Jaqueline Molina Barrios2, Madelyne Geovanna Moya Trejo3, Geovanna Michele Moya Díaz4.
1Estudiante de V año de medicina, Universidad Católica de Honduras, UNICAH.
2Médico general con maestría en psicología educativa, Docente de embriología, UNICAH.
3Estudiante de V año de medicina, UNICAH.
4Médico general de la Universidad Nacional Autónoma de Honduras, Bioeticista FLACSO Argentina.
Correspondencia a: Steven Josue Navarro Turcios.
Correo electrónico: steven13navarro@gmail.com
Telefono: +504 8831-0240
Procedencia y arbitraje: no comisionado, sometido a arbitraje externo.
Recibido para publicación: 10 de mayo de 2020
Aceptado para publicación: 4 de enero de 2021
Citar como: Navarro Turcios SJ, Molina Barrios SJ, Moya Trejo MG, Moya Díaz GM. Reporte de casos de ictiosis laminar en honduras; desafío diagnóstico. Rev Cient Cienc Med. 2020; 23(2): 271-277
RESUMEN
Introducción: La ictiosis es una enfermedad de origen genético que afecta principalmente la piel, posee varios tipos dependiendo el patrón de herencia. Dentro de las dominantes encontramos a la ictiosis vulgar, y en las recesivas la Ictiosis laminar, Eritroderma ictiosiforme, arlequín.
La ictiosis laminar es una genodermatitis congénita que según datos estadísticos internacionales (EUA) reporta 1:200 000-300 000 recién nacidos, dentro de las bases de datos consultadas en Honduras, no se encontró la incidencia y prevalencia de esta enfermedad.
Materiales y métodos: Se analizó el árbol genealógico de 6 pacientes más la valoración de las manifestaciones clínicas propias de cada individuo para determinar el patrón de herencia especifico y así identificar el tipo de ictiosis.
Resultado: El análisis mostró un patrón de herencia autosómico recesivo, consanguinidad en padres de los pacientes.
Conclusiones: La realización de un pedigríes esencial en conjunto con la exploración clínica para el diagnóstico de ictiosis, en ausencia de pruebas genéticas e histopatológicas.
Palabras clave: Ectropión, ictiosis lamelar, queratodermia palmoplantar, Honduras.
ABSTRACT
Introduction: Ichthyosis is a disease of genetic origin in which the skin is mainly affected, it has different types depending on the inheritance pattern. Within the dominants we find the ichthyosis vulgaris and on the recessive ones we have the lamellar ichthyosis, harlequin Ichthyosis and Ichthyosiform erythroderma.
Lamellar Ichthyosis is a congenital genodermatitis that according to international statistical data (US) it reports 1:200 000-300 000 newborns. Within the databases consulted in Honduras no prevalence and incidence of the disease was found.
Materials and methods: It was analyzed the genealogical tree of 6 patients plus the valuation of clinical manifestations of each individual to determine the specific inheritance pattern to identify the type of Ichthyosis.
Results: The analysis demonstrated an autosomal inheritance pattern and consanguinity in the patient's parents.
Conclusions: The realization of a pedigree it's essential in conjunction with clinical examination forthe diagnosis of ichthyosis in absence of histopathologic and genetical tests.
Keywords: Ectropion, Ichthyosis, Lamellar, Keratoderma, Palmoplantar, Honduras.
INTRODUCCIÓN
La ictiosis congénita es una enfermedad de origen genético cuya afección principal es en la piel 'genodermatitis', produce un trastorno que conlleva a la hiperqueratinización heterogénea y generalizada manifestando en el individuo sequedad y descamación continua del órgano.1,2
Según los resultados de la primera conferencia del consenso de ictiosis llevada a cabo en el 2009, citada por Rodríguez, Ginarte, Vega y Toribio en el 20133 y Takeichiy Akiyama en el 2016,4 existen múltiples clasificaciones, sin embargo, la de mayor relevancia en la práctica clínica es según, el patrón de herencia que las clasifica en; ictiosis autosómicas dominantes (vulgar y ligada al cromosoma X) y las autosómicas recesivas (laminar, eritroderma ictiosiforme congénita bullosa y no bullosa e ictiosis arlequín), siendo la última mencionada la más agresiva y de peor pronóstico.3,4
La ictiosis laminar (IL) es la variante más frecuente de las ictiosis congénitas que se transmiten de forma autosómica recesiva, este tipo se caracteriza por una presentación especial del producto al momento del nacimiento denominada 'bebé colodión', el recién nacido muestra una membrana traslúcida que rodea la totalidad de la piel.5
Según datos estadísticos internacionales (EEUU) la IL afecta1; 200 000-300 000 recién nacidos,1,3,6 dentro de las bases de literatura nacionales que se revisaron no se encontraron datos epidemiológicos que muestren la prevalencia e incidencia real de la enfermedad, sin embargo, existen casos publicados de; ictiosis arlequín por Ávila et al en el 2011, eritroderma ictiosiforme por Valladares etal en el 20179 y Navarro et al en el 2018.8
En Honduras actualmente no se realizan pruebas genéticas y moleculares para confirmar el diagnóstico, por tal motivo es de vital importancia la realización de un pedigrí en las patologías de origen genético más la correlación de signos y síntomas encontrados en los pacientes, y de esta forma realizar un diagnóstico clínico apropiado.
La presente serie de casos se realizó en la aldea de Trinidad de quebradas, municipio de Vallecillo, departamento de Francisco Morazán, Honduras. En dicho lugar actualmente se reportan 9 casos clínicamente confirmados de ictiosis congénita entre adultos hombres, mujeres y niños. Se realizó el proceso de consentimiento informado brindando la información pertinente a los pacientes, respetando su autonomía y garantizando la confidencialidad de su información personal. Seis pacientes aceptaron participar. Este estudio tiene como objetivo el análisis de un árbol genealógico más la valoración de las manifestaciones clínicas propias de cada individuo para determinar el patrón de herencia específico y así identificar el tipo de ictiosis.
PRESENTACIÓN DEL CASO
La serie de casos que a continuación se describe, consta de seis pacientes con diagnósticos clínicos confirmados, todos originarios de una misma región. Mediante el estudio del árbol genealógico (ver Fig. 1, I generación) se determinó que los seis individuos poseen un parentesco familiar, información que todos desconocían, únicamente los casos #4, 5, 6, refirieron ser parientes cercanos y son los únicos que reciben atención médica en la actualidad.

La evaluación clínica individual de los pacientes se realizó en sus domicilios, debido al difícil acceso a los servicios de salud en su comunidad y para su propia comodidad. Todos los casos al momento de su nacimiento se manifestaron como bebés colodión, considerado un signo fenotípico característico de la ictiosis laminar. Posteriormente desarrollaron la presentación clínica de este tipo de ictiosis, que incluye: escamas (placas hiperqueratosicas) de color marrón negruzco, xerosis, hipohidrosis o anhidrosis, queratodermia palmo-plantar, alopecia cicatrizal y eversión bilateral de párpado (ectropión). Cada uno de los signos mencionados serán descritos individualmente en cada uno de los pacientes:
Caso. 1
Individuo (IND) n1- V generación (GN) (ver Fig. 1).
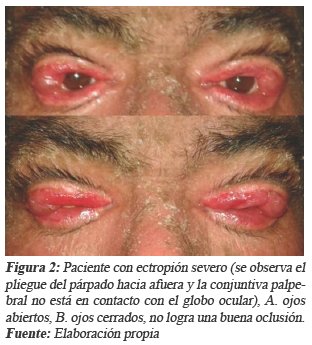
Masculino de 45 años, al examen físico presentó ectropión severo, con presencia de lagoftalmos que impide apreciar reflejo de bell, lagrimeo e irritación, extensión de mucosa hasta la conjuntiva, afectando la visión por cubrir parte del iris y la pupila (ver Fig. 2).
Caso. 2
IND. n6-V GN(ver Fig. 1).
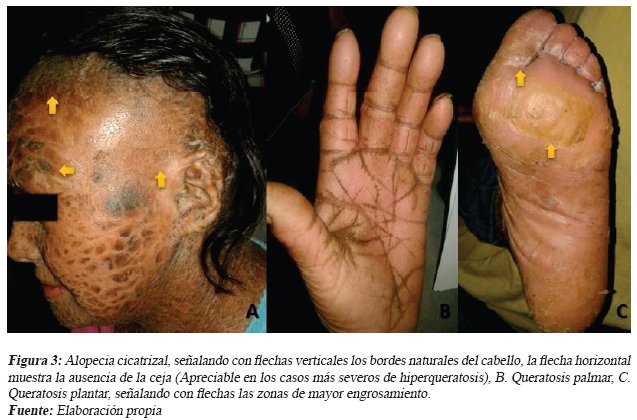
Femenina de 41 años, al examen físico presentó hipoplasia del cartílago auricular y alopecia cicatrizal acompañada por atrofia severa de los folículos pilosos con reemplazo irreversible del tejido fibroso, existe descamación activa del cuero cabelludo con predominio en la región frontal (ver Fig. 3. A).
Caso. 3
IND. n7- V GN, hijo de familiares en cuarto grado de consanguinidad (ver Fig. 1).
Masculino mayor de edad, al examen físico presentó severa hiperqueratosis palmo-plantar con pliegues palmares acantósicos pronunciados y placas heterogéneas, con bordes indefinidos en la región plantar (ver Fig. 3. B-C).
Caso. 4
IND. #1-VII GN, hija de padres consanguíneos en tercer grado (Ver Fig. 1).
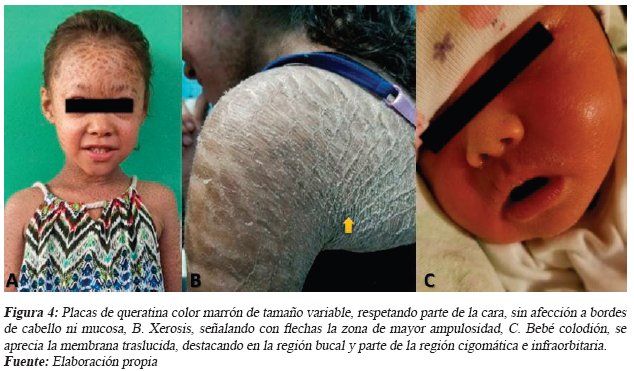
Escolar femenina de 7 años, al examen físico presentó múltiples placas hiperqueratosicas, simétricas, color marrón oscuro, formando un patrón similar a una red, de tamaño variable, con bordes delimitados, textura indurada con leve resistencia al tacto, afectando la mayor parte del cuerpo, disminuyendo su intensidad en la mitad inferior de la cara y flexuras anatómicas (ver Fig. 4. A).
Caso. 5
IND. n2- VII GN, familiar en segundo grado de consanguinidad con la paciente n4 (Ver Fig. 1).
Adolescente femenina de 15 años, al examen físico se aprecia xerosis que se distingue por la acentuación de marcas con pliegues longitudinales en el área axilar acompañado de escamacion gruesa e hiperpigmentación (ver Fig. 4. B), hipohidrosis, leve hiperqueratosis palmo-plantar.
Caso. 6
IND. n5- VII GN, familiar en primer grado de consanguinidad con la paciente n5 (Ver Fig. 1).
Preescolar femenina de 3 años, presentó clínica de ictiosis laminar a la evaluación. Madre de paciente refiere presentación como bebé colodión en el nacimiento, manifestando una membrana corporal gruesa, tensa y brillante, leve ectropión y eclabión. Tambien relata el desprendimiento de la membrana entre la primera y cuarta semana de vida, posterior a este evento evidenciando el fenotipo definitivo de ictiosis laminar (ver Fig. 4. C).
DISCUSIÓN
La ictiosis laminar (IL) es una enfermedad congénita rara, su nombre deriva del griego 'ichtys' que significa pez, conocida también como la 'enfermedad de la piel de pescado'12. Se caracteriza por producir una acumulación excesiva de grandes cantidades de escamas (cornificación) en la capa superior de la piel y no hay discriminación de sexo en los afectados, 10,11. Dichas características fueron evidentes en todos los pacientes anteriormente descritos.
Muchos autores han determinado que la IL se transmite de forma autosómica recesiva 1,8,13. Esto se evidencia a través del árbol genealógico presentado (Fig. 1), donde se observan relaciones consanguíneas en los individuos (IND) #9 y 11- IV generación (GN) de IND #1 y 2-VI GN. También se aprecia que los casos confirmados únicamente están en la V y VII GN, identificando una transmisión horizontal de la patología (presencia de saltos generacionales).
En el pedigrí como parte del estudio genético, se aprecia que toda la estirpe parte de tres familiares en primer grado, IND #1, 3, 5 de la I GN, posteriormente cada uno tiene su propia descendencia. Hay que tomar en consideración que todas las personas son originarias de la misma comunidad donde existe endogamia, factor que predispone a confirmar el patrón de herencia autosómico recesivo12,14,15,26.
Con mayor frecuencia la IL y eritroderma ictiosiforme congénita bullosa/no bullosa (EICB/NB), con la modalidad de herencia previamente descrita, son evidentes en el nacimiento, motivo por el cual se les denomina ictiosis verdaderas13,16,17. Ambas se caracterizan por presentarse como 'Bebé Colodión' en el momento del parto, caracterizado por poseer una membrana traslucida que recubre la totalidad de la piel, dando un aspecto brillante, presentando algunas complicaciones como limitación de la movilidad, eclabión y ectropión5, 12,13,16,18,26. Condición que presentaron todos los pacientes descritos al nacer.
El diagnóstico clínico diferencial de la IL es la EICB/NB, esta última se diferencia por el predominio de un cuadro eritematoso, 9,12,19. En cambio en los pacientes con IL este signo es ausente o mínimos, poseen escamas en formas de placa, que tienen un color oscuro y cubren la mayor parte o totalidad del cuerpo, descamación activa de la piel, distintos grados de resequedad de la piel o xerosis y disminución de sudoración (Hipohidrosis) que puede llegar hasta la ausencia (Anhidrosis), esto como resultado de la obstrucción de las glándulas sudoríparas 3,5,8,20. Basándonos en la ausencia de la eritroderma más las características clínicas ya expuestas en la literatura se confirmó en estos pacientes el diagnóstico clínico de ictiosis laminar.
Las complicaciones de la enfermedad varia del grado de severidad de la hiperqueratosis, suele ser manifiesto por los pacientes que han recibido mínimo o nulo tratamiento médico, según un estudio retrospectivo realizado en Argentina en el 2015, menciona que la complicación oftalmológica más frecuente es el ectropión (80% de pacientes con ictiosis lamelar), también se puede observar lagoftalmos e hiperemia conjuntival,21 en algunas ocasiones eclabión, alopecia cicatrizal especialmente en bordes del cuero cabelludo, y la queratinización excesiva y anómala de las palmas de las manos y plantas de los pies (Queratodermia o hiperqueratosis palmo-plantar)3,12,13 Datos congruentes con las complicaciones de los pacientes que no reciben tratamiento.
El Gold standard para el diagnóstico y tipificación de la ictiosis es el estudio de los genes mutados, de los cuales se han identificado nueve por causar formas no sindrómicas de las ictiosis congénitas autosómicas recesivas 22,23. Tales genes son ALOX12B; ALOXE3; ACBA12; CERS3; CYP4F22; NIPAL4/ICHTHYIN; PNPLA1; SDR9C7 y TGM1 siendo este último el gen más comúnmente detectado en la ictiosis laminar3,4. La función del gen TGM1 es codificar la enzima TGAsa 1, una de las tres enzimas encontradas en la epidermis20 . En los pacientes descritos en la serie de casos, no fue posible realizarles estudio genético ya que no está disponible en el país.
Otro método útil para el diagnóstico de la ictiosis, son los análisis histopatológicos, varios estudios demuestran que en la IL se observa una masiva hiperqueratosis ortoqueratótica (queratocitos sin núcleo), La epidermis se encuentra acantosica y ocasionalmente adquiere una apariencia similar a la psoriasis, la velocidad de proliferación celular es normal o ligeramente elevada 24. En esta serie de casos no fue posible realizar estudios histológicos debido al difícil acceso a los servicios de salud donde se realizan exámenes complejos como estos. Sin embargo, a pesar de que los estudios histológicos han sido útiles para el diagnóstico, su aplicación no siempre permite diferenciar los tipos de ictiosis, razón por la que es recomendable siempre establecer el patrón de herencia para ratificar el tipo sospechado 25,26.
Finalmente, aunque ya se discutieron las formas más precisas de diagnosticar y clasificar la ictiosis laminar en un sistema de salud ideal, y considerando la limitación que enfrentan estos pacientes para realizarse estudios genéticos e histológicos en el país, se hizo énfasis en el análisis del patrón de herencia, que en conjunto con la exploración clínica permite lograr un diagnóstico apropiado de la enfermedad.
La realización de un árbol genealógico es fundamental en las patologías de origen genético para el reconocimiento de un patrón de herencia especifico. Por lo que se recomienda que debe incluirse como parte de la evaluación personalizada de cada paciente con sospecha de ictiosis.
Así mismo es importante brindar asesoría a las comunidades donde existen casos confirmados de ictiosis, para evitar la endogamia y las repercusiones de esta enfermedad.
Agradecimientos
Se agradece el apoyo por sus asesorías y recomendaciones a la Dra. Evelyn Paola Matamoros Valle, Médico general de la Universidad Nacional Autónoma de Honduras y al Dr. Ramón Yefrin Maradiaga Montoya, Médico internista y docente de fisiopatología y semiología de la Universidad Católica de Honduras.
REFERENCIAS
1. Osório F,LeãO M ,Azevedo F,Magina S. Lamellar Ichthyosis due to ALOX12B mutation. Elsevier[internet].2013[citado 5 Enero 2020];104(5):443-444. Disponible en: https://doi.org/10.1016/j.adengl.2012.07.026
2. Leight H,Zinn Z,Jalali O. Bilateral lower extremity hyperkeratotic plaques: a case report of Ichthyosis vulgaris. CCID[internet].2015[citado 3 Mayo 2020];2015:(8)485-488. Disponible en: https://doi.org/10.2147/CCID.S89871
3. Rodriguez-Pazos L,Ginarte M,Vega A,Toribio J. Autosomal Recessive Congenital Ichthyosis. AD[internet].2013[citado 3 Mayo 2020];104(4):270-284. Disponible en: https://www.actasdermo.org/es-ictiosis-congenitas-autosomicas-recesivas-articulo-S0001731012001998
4. Takeichi T,Akiyama M. Inherited Ichthyosis:Non-syndromic forms. TJD[internet].2016[citado 24 Abril 2020];43:242-251. Disponible en: https://onlinelibrary.wiley.com/doi/epdf/10.1111/1346-8138.13243
5. De Leonibus,Lembo C,Santantonio S,Fioretti T,Rojo S,Salvatore F,et al. Lamellar Ichthyosis and arthrogryposis in a premature neonate. JDCR[internet].2015[citado 3 Mayo 2020];9(2):49-51.disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4517804/
6. Binamer Y. Ichthyin (NIPAL4)-autosomal recessive congenital ichthyosis with atopic diathesis:Case report and literature review. JDDS[internet].2015[citado 4 Mayo 2020];20(2016):55-57. Disponible en: http://dx.doi.org/10.1016/j.jdds.2015.07.002
7. Ávila Montes G, Montoya M,Bustillo M,Carrasco D,Rodriguez Maradiaga C, Carranza N. Ictiosis Arlequín:reporte de un caso en Honduras y revisión de la literatura. RMH[internet]. 2011[citado 4 Mayo 2020];79(4):199-202. Disponible en: http://www.bvs.hn/RMH/pdf/2011/pdf/Vol79-4-2011-7.pdf
8. Navarro S,Molina Barrios S. Caso de Ictiosis Laminar en Honduras. RCCM [internet].2018[citado 4 Mayo 2020];21(2):73-74. Disponible en: http://www.scielo.org.bo/scielo.php?script=sci_arttext&pid=S1817-74332018000200013
9. Valladares Ramos M, Caballero Castro H. Eritroderma Ictiosiforme congénita no ampollosa, APH[internet].2017[citado 17 Septiembre 2019];8(1): 751. Disponible en: https://www.camjol.info/index.php/PEDIATRICA/article/view/7594
10. Del Catillo Ruiz V, Uranga R, De la Rosa G. Genética Clínica. 2nd ed. Ciudad de México: Manual Moderno (Apoyo Multimedia); 2019.
11. Ictiosis lamenar [Internet]. Federación española de enfermedades raras. 2020 [cited 6 May 2020]. Available from: https://enfermedades-raras.org/index.php/component/content/article?id=3100&idpat=209.
12. Martínez T. Información sobre la Ictiosis. ASIC[Internet].2020 [citado 6 Mayo 2020].Disponible en: https://www.ictiosis.org/sobre_ictiosis/Info_ictiosis/info.htm.
13. Morales Barrera Ma. E, Zabala Martínez M. Ictiosis laminar; Caso clínico. RCDP[Internet].2011[citado 6 Mayo 2020];20(1):5-8. Disponible en: https://www.medigraphic.com/cgi-bin/new/publicaciones.cgi?IDREVISTA=34&NOMBRE=Revista%2520%2520del%2520Centro%2520Dermatol%25F3.
14. Suárez M, Lozano M. Patrones de herencia [Internet]. Medicos.cr. [citado 7 Mayo 2020]. Disponible en: http://www.medicos.cr/web/documentos/EMC%202015/charlasmedicinamolecular/Patrones%20de%20Herencia.pdf
15. Dallaire L, Huret J, Vizmanos J. Patrones mendelianos y atípicos de herencia [Internet]. Atlas of Genetics and Cytogenetics in Oncology and Haematology. 2002 [citado 7 Mayo 2020]. Disponible en: http://atlasgeneticsoncology.org/Educ/GenetFormelSpaID30025SS.html
16. Estrada López A, García Miranda G, Meza-Zamora D. Bebé colodión; Reporte de caso. Medigraphic [Internet]. 2010 [citado 7 Mayo 2020];67(4):352-358. Disponible en: http://www.scielo.org.mx/pdf/bmim/v67n4/v67n4a7.pdf
17. Scacchi MF, Pagotto B, Correa N, Castillo A, Luna PC, Boggio P. Bebé colodión. Comunicación de 14 casos. Dermatol Argent [Internet]. 2011 [citado 07 Mayo 2020];17(2):128-33. Disponible en: http://www.dermatolarg.org.ar/index.php/dermatolarg/article/download/621/395
18. Sotolongo Díaz D, Pérez Jiménez A, Obregón Valdivia P, Pérez de Corcho IM, Navarro Pírez I. Bebé colodión siete años después. presentación de caso. medigraphic [Internet]. 2015 [citado 7 Mayo 2020];21(3):51-54. Disponible en: https://www.medigraphic.com/pdfs/mediciego/mdc-2015/mdc153g.pdf
19. Sánchez Linares V, García González A, Nazco Fariña G, Gómez Díaz Y, Márquez del Pozo N. Eritrodermia ictiosiforme congénita no ampollosa asociada a pénfigo vegetante. Presentación de caso. GME [Internet]. 2016 [citado 7 Mayo 2020];18(1). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1608-89212016000100010
20. Gulasi S. Congenital Ichthyosis: a case treated successfully with acitretin. IJP [internet]2016[citado Mayo 5 2020]; 5:1-3. Disponible en: http://ijp.tums.pub/articles/2442.html
21. Puccio M, Forniés Paz M, Polo C, López B, Fandiño A. ICTIOSIS CONGENITA: Manifestaciones oftalmológicas. RMI[Internet]. 2015 [citado 7 Mayo 2020];22(2):116-119. Disponible en: http://www.medicinainfantil.org.ar/images/stories/volumen/2015/xxii_2_116.pdf
22. Heinz L, Kim G-J, Marrakchi S, Christiansen J, Turki H, Rauschendorf M-A, Lathrop M, Hausser I, Zimmer A-D, Fischer J. Mutations in SULT2B1 Cause Autosomal-Recessive Congenital Ichthyosis in Humans. TAJHG[internet]2017[citado 6 Mayo 2020];100:926-939. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5473727/pdf/main.pdf
23. Sugiura K,Akiyama M. Lamellar Ichthyosis Caused by a Previously Unreported Homozygous ALOXE3 Mutation in East Asia. ADV[internet].2015[citado 25 Abril 2020];95:858-859. Disponible en: https://www.medicaljournals.se/acta/content_files/files/pdf/95/7/4324.pdf
24. Ozge Mine O,Funda T,Evren S,Umran Y. Lamellar ichthyosis-like eruption associated with ponatinib. AD APA [internet].2016[citado 5 Mayo 2020];25:59-60. Disponible en: http://acta-apa.mf.uni-lj.si/journals/acta-dermatovenerol-apa/papers/10.15570/actaapa.2016.17/actaapa.2016.17.pdf
25. Leight H, Zinn Z, Jalali O. Bilateral lower extremity hyperkeratotic plaques: a case report of ichthyosis vulgaris. Clinical, Cosmetic and Investigational Dermatology [Internet]. 2015 [cited 18 January 2020]; 8:485–488. Available from: https://www.dovepress.com/bilateral-lower-extremity-hyperkeratotic-plaques-a-case-report-of-icht-peer-reviewed-article-CCID
26. Nashan Dortmund D. Ichthyoses in everyday practice: management of a rare group of diseases. Journal of the Germany Society of Dermatology [Internet]. 2020 [citado 7 mayo 2020];0: 225–243. Disponible en: https://onlinelibrary.wiley.com/doi/epdf/10.1111/ddg.14049

