










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Científica Ciencia Médica
Print version ISSN 1817-7433On-line version ISSN 2220-2234
Rev Cient Cienc Méd vol.23 no.2 Cochabamba 2020
CASO CLÍNICO
COMPROMISO CARDIOVASCULAR EN EL SÍNDROME DE MARFAN: REPORTE DE CASO
CARDIOVASCULAR COMPROMISE IN MARFAN SYNDROME: CASE REPORT
Andree J. Michel Martínez 1![]() , Roberto Joaquín Chavez Camacho 1
, Roberto Joaquín Chavez Camacho 1![]() , Roy Arnold Laguna Chavez2
, Roy Arnold Laguna Chavez2
1Interno de Medicina de la Facultad de Medicina de la Universidad Mayor de San Andrés, La Paz - Bolivia.
2Interno de Medicina de la Facultad de Medicina de la Universidad Franz Tamayo
Correspondencia a: Andree J. Michel-Martínez
Correo electrónico: a.n.d.r.e.e@hotmail.com
Telefono: +591 71574469
ORCID: https://orcid.org/0000-0002-2443-0932
https://orcid.org/0000-0001-9154-3577
Procedencia y arbitraje: no comisionado, sometido a arbitraje externo.
Recibido para publicación: 16 de abril de 2020
Aceptado para publicación: 18 de enero de 2021
Citar como: Michel-Martínez AJ, Chavez-Camacho RJ, Laguna-Chavez RA. Compromiso cardiovascular en el síndrome de marfan: reporte de caso: Rev Cient Cienc Med. 2020; 23(2): 252-257
RESUMEN
El Síndrome de Marfanes una enfermedad del tejido conectivo causada por mutaciones en el gen FBN I,mismo que codifica la fibrilina-1, glucoproteína fundamental del componente de las microfibrillas. Entre las manifestaciones clínicas, la afectación cardiovascular merece una consideración especial, debido a su pronóstico. Se presenta un varón de 40 años quien acude al Instituto Nacional del Tórax por un cuadro clínico de 2 años de evolución caracterizado por clínica de insuficiencia cardiaca descompensada, insuficiencia aortica y criterios colagenopatía subyacente, ante lo cual tras los estudios de gabinete y el uso de los criterios pertinentes (Ghent modificados) se llega al diagnóstico de Síndrome de Marfan. La supervivencia de estos pacientes depende del diagnóstico temprano evitando las complicaciones que en su mayoría son mortales, el uso de los scores es de ayuda y la intervención oportuna lleva a un mejor pronóstico de vida y evita los procedimientos invasivos y por ende demás complicaciones.
Palabras clave: Síndrome de Marfan, fibrilina-1, insuficiencia cardiaca
ABSTRACT
Marfan syndrome is a connective tissue disease caused by mutations in the FBN I gene, which encodes fibrillin-1, a fundamental glycoprotein of the microfibril component. Among the clinical manifestations, cardiovascular involvement deserves special consideration, due to its prognosis. We present a 40-year-old man who came to the National Thorax Institute for a clinical picture of 2 years of evolution characterized by symptoms of decompensated heart failure, aortic insufficiency and underlying collagenous criteria, before which, after cabinet studies and the use of the relevant criteria (modified Ghent) leads to the diagnosis of Marfan Syndrome. The survival of these patients depends on early diagnosis, avoiding complications that are mostly fatal, the use of scores is helpful and timely intervention leads to a better prognosis for life and avoids invasive procedures and therefore other complications.
Keywords: Marfan syndrome, fibrillin-1, heart failure
INTRODUCCIÓN
El Síndrome de Marfan (SM) es enfermedad del colágeno hereditarias, su etiología se debe a mutaciones heterocigóticas en el gen FBN1 (localizado en el cromosoma 15q21.1) que codifica la fibrilina 1, componente de la red de microfibrillas que sirve de armazón para el depósito de elastina y el ensamblaje de las fibras elásticas, misma que al verse afectada en su síntesis conduce a la debilidad del tejido, aumento de TGF-β, pérdida de las interacciones célula-matriz, dando lugar a las diferentes manifestaciones fenotípicas. 1,2,7
Su incidencia aproximada es de 1 por cada 5.000 a 1-3 por cada 10.000 personas vivas, según las series, afecta por igual a ambos sexos y su distribución es mundial.1,3
Es relevante tener en cuenta que este es un trastorno multisistémico con manifestaciones que típicamente involucran el sistema cardiovascular, sistemas esqueléticos y oculares; de estas las manifestaciones cardiovasculares son detectables en el examen físico en 80% de los casos, siendo muy sutiles en la infancia y causa de morbimortalidad en 90% de pacientes, principalmente por enfermedad aórtica y sus complicaciones por lo cual amerita darles una consideración especial debido a su impacto en el pronóstico de vida del paciente.4,7,9
El diagnóstico se basa en los signos clínicos multisistémicos y en la historia familiar englobados en la actualidad en los criterios diagnósticos de Ghent modificados.1
PRESENTACIÓN DEL CASO
Paciente masculino de 40 años de edad procedente y residente de Juliaca-Perú acude al Hospital Carlos Monge Medrano de su localidad con un cuadro clínico caracterizado por presentar aumento de volumen en extremidades inferiores, simétrico, bilateral y ascendente hasta el tercio medio de ambas extremidades, además de disnea de medianos a pequeños esfuerzos y palpitaciones asociadas a tos seca sin predominio horario. Tras su evaluación se le instaura tratamiento a base de diuréticos (no menciona nombre); al no encontrar mejoría el paciente por recomendación de su familia decide emprender un viaje para consultar en el Instituto Nacional Del Tórax (INT) en el país de Bolivia.
El mismo acude al Servicio de Emergencias del INT con mismo cuadro más con una evolución de 6 meses. En sus antecedentes el paciente refiere: en 2011 fue diagnosticado y tratado quirúrgicamente de Hernia Inguinal derecha no complicada en el Hospital de Clínicas, en 2016 fue diagnosticado de Insuficiencia Cardiaca por coartación de aorta por ecocardiografía transtorácica misma que fue tratada con diuréticos (no refiere nombre) y no refiere familiares con misma patología.
Se procede a su evaluación por emergencias y se encuentran los siguientes hallazgos positivos en el examen físico:
Al examen físico se encuentra signos vitales de PA 100/30 mmHg; FC 82 lpm; FR 26 rpm; T° 36,2 C; peso 65 kg; talla 1,65 m; IMC 24 m2/kg. Llama la atención en cabeza estrabismo convergente derecho; luxación de cristalino izquierdo; agudeza visual severamente disminuida en ojo derecho; paladar ojival; signo de muller(+); cuello: ingurgitación yugular (++); tórax: a la inspección en quilla; a la auscultación ritmo de galope, soplo sistólico (III/VI) en área aórtica, soplo diastólico (III/VI) en área mitral (Austin Flint), pulmonar: estertores sub-crepitantes en ambas regiones subescapulares; extremidades: aracnodactilia; hiperlaxitud articular; signo de quincke (+); pie plano; relación miembros superiores y altura > 1. (véase figura 1)
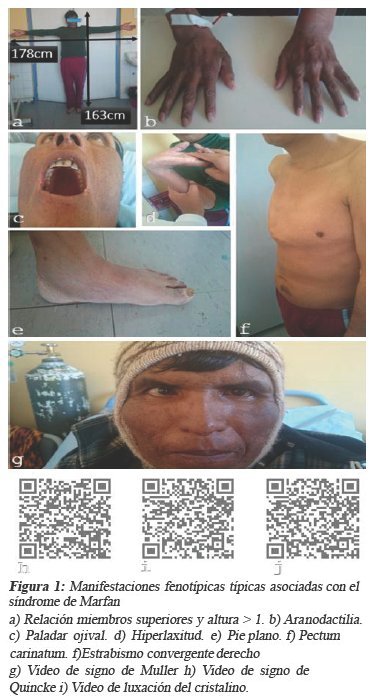
Se decide su internación en el servicio de cardiología del INT y se piden examines complementarios donde se encuentran: El electrocardiograma con ritmo sinusal, FC 65 lpm eje eléctrico de QRS en +60°; índice de cornell +; onda p bimodal en DI y DII (crecimiento auricular izquierdo); + - - en V1 (sobrecarga de aurícula izquierda); sobrecarga sistólica del ventrículo izquierdo. (Figura 2).
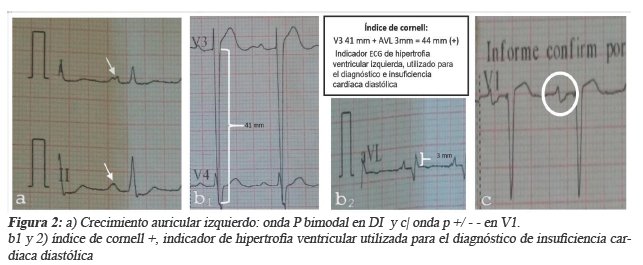
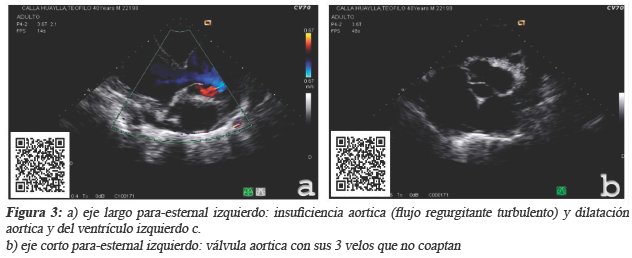
Los laboratorios con Creatinina 1,8 mg/dl,TSH60uUI/ml(0,4-4), TT3 y TT4 normales (hipotiroidismo subclínico (véase figura 2). La ecocardiografía muestra insuficiencia aórtica con dilatación de aorta ascendente, dilatación severa de ambas aurículas, dilatación severa de ventrículo izquierdo con función sistólica global severamente disminuido, dilatación moderada de ventrículo derecho con disfunción sistólica leve, insuficiencia pulmonar mínima, insuficiencia mitral leve, insuficiencia tricuspídea leve, hipertensión arterial pulmonar leve con resistencias vasculares. (Ver Figura 3)
Con todo lo anterior se llega a los siguientes diagnósticos: Insuficiencia aórtica severa con dilatación de aorta ascendente secundaria a Síndrome de Marfan, Hipertensión arterial pulmonar tipo 2 NIZZA, Insuficiencia cardíaca congestiva CF II según la NYHA e Hipotiroidismo subclínico en tratamiento.
Se inician las indicaciones de la tabla 1, de las cuales todas se cumplieron a excepción del cambio valvular aórtico quirúrgico por motivos económicos del paciente.

En buena respuesta al tratamiento tras 2 semanas de internación paciente ya en mejores condiciones generales se decide alta hospitalaria y se le indica al paciente seguir con la espironolactona, furosemida, enalapril y además adherir el bisoprolol a dosis baja (1,25 mg de bisoprolol una vez al día durante una semana) e incrementarla gradualmente esto bajo control por consulta externa.
DISCUSIÓN
El SM es una enfermedad genética autosómico dominante, que se caracteriza por un compromiso del tejido conectivo, debido a una mutación a nivel del gen de fibrina 1, causando un alto de grado de compromiso patológico a nivel de todos los sistemas.11
La principal complicación es la dilatación de la raíz aórtica es la manifestación cardiovascular más común que ocurre en 60 a 80% de los pacientes y causa mortalidad en 90% de pacientes por lo cual un diagnóstico oportuno es necesario.3,10,11
El diagnóstico del SM es actualmente clínico en el cual se basa en los hallazgos en diferentes órganos y sistemas, y tomando en cuenta la historia familiar del paciente, el diagnóstico de estos pacientes se abordó por primera vez a través de los criterios diagnósticos Ghent para el SM que se remontan a 1996, todos estos fueron modificados con el tiempo ya que muchas personas diagnosticadas con el trastorno no portaban la mutación FBN1 identificada en miembros de la familia más severamente afectados, esto llevó a la creación de nuevos criterios modificados y revisados (véase la tabla 2).4,5
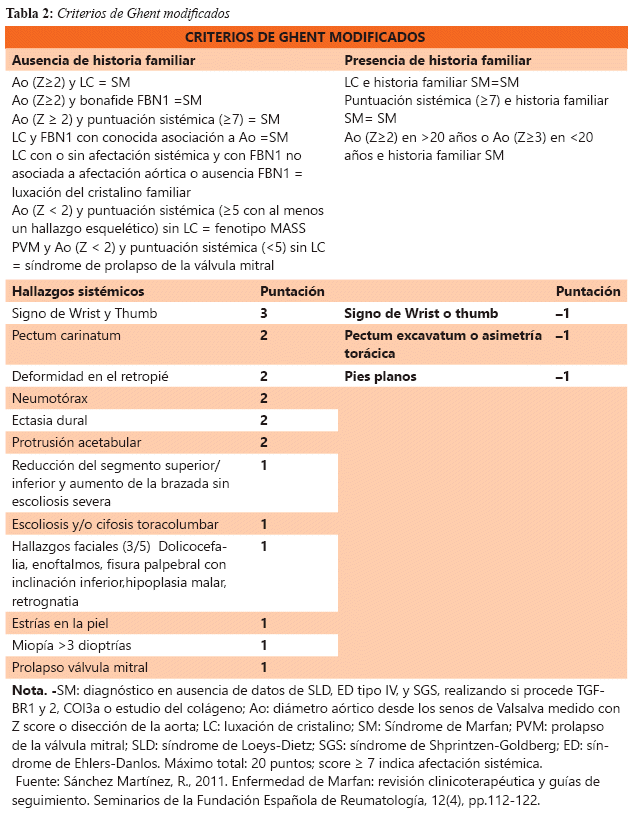
En el caso de nuestro paciente el debutó con el cuadro de insuficiencia cardiaca descompensada a múltiples patologías cardiacas en la cual resalta la típica dilatación aórtica que según el Score-Z obtenemos un 8.3 mismo que es mayor a 2 con lo cual ya contamos con un criterio mayor. En el examen físico el paciente presenta Signo de Wrist y Thumb, Pectum carinatum, pies planos reducción del segmento superior/inferior y aumento de la brazada sin escoliosis mismo que nos arroja un puntaje de 7 con el cual ya podemos ver que tiene una afectación sistémica, por lo cual requiere manejo multidisciplinario.1,2
En nuestro paciente el inicio del tratamiento se indicó diuréticos, para la insuficiencia cardiaca descompensada, posteriormente se inicia bloqueadores beta (bisoprolol) según directrices de la aorta torácica ACC / AHA / AATS de 2010 esto para reducir la tasa de dilatación aortica.2,6
Conforme al tratamiento quirúrgico se recomienda al paciente cirugía profiláctica de la raíz aórtica estrategia para prevenir la disección de aorta ya que a fines de la década de 1960 del procedimiento Bentall, logra una supervivencia operativa superior al 98%. La cual es rechazada por motivos económicos.2,10
CONCLUSIÓN
En todo lo expuesto sobre el SM, el diagnóstico oportuno es lo más importante, ya que a edad más tardía aumenta las complicaciones cardiovasculares con una mortalidad del 90%. El diagnóstico genético por mutaciones del gen fibrina 1 queda sin importancia al momento de realizar el diagnóstico, ya que es clínico.
El conocimiento adquirido a través de la investigación, centrada en el SM ha mejorado y continuará mejorando la atención de los pacientes.
REFERENCIAS
1. Isekame Y, Gati S, Aragon-Martin J, Bastiaenen R, Kondapally Seshasai S, Child A. Cardiovascular Management of Adults with Marfan Syndrome. European Cardiology Review. [Internet] 2016 [Consultado 2 febrero de 2020];11(2):102. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6159454/
2. Judge D, Dietz H. Marfan's syndrome. The Lancet. [Internet] 2005 [Consultado 2 febrero de 2020];366(9501):1965-1976. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/16325700
3. Aoyama T, Francke U, Dietz H, Furthmayr H. Quantitative differences in biosynthesis and extracellular deposition of fibrillin in cultured fibroblasts distinguish five groups of Marfan syndrome patients and suggest distinct pathogenetic mechanisms. Journal of Clinical Investigation. [Internet] 1994 [Consultado 2 febrero de 2020] ;94(1):130-137. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC296290/pdf/jcinvest00019-0148.pdf
4. Lebreiro A, Martins E, Cruz C, Almeida J, Maciel M, Cardoso J. Marfan syndrome: clinical manifestations, pathophysiology and new outlook on drug therapy. Rev Port Cardiol [Internet]. 2010 [Consultado 2 Febrero 2020]; (29):1021-36. Disponible en: https://www.researchgate.net/publication/47517934_Marfan_syndrome_Clinical_manifestations_pathophysiology_and_new_outlook_on_drug_therapy
5. Martín C, Evangelista A, Serrano-Fiz S, Villar S, Ospina V, Martínez D et al. Aortic Complications in Marfan Syndrome: Should We Anticipate Preventive Aortic Root Surgery?. The Annals of Thoracic Surgery. [Internet] 2019 [Consultado 24 febrero de 2020];129 (98):123-129 Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/31589859
6. Cañadas V, Vilacosta I, Bruna I, Fuster V. Marfan syndrome. Part 1: pathophysiology and diagnosis. Nature Reviews Cardiology. [Internet] 2010 [Consultado 2 febrero de 2020];7(5):256-265.Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/20351703
7. Pearson G, Devereux R, Loeys B, Maslen C, Milewicz D, Pyeritz R et al. Report of the National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group on Research in Marfan Syndrome and Related Disorders. Circulation. [Internet] 2008 [Consultado 30 febrero de 2020];118(7):785-791 Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/18695204
8. Sakai L, Keene D, Renard M, De Backer J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene. [Internet] 2016 [Consultado 28 febrero de 2020];591(1):279-291. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6639799/
9. Wright M, Connolly H. Genetics, clinical features, and diagnosis of Marfan syndrome and related disorders. UpToDate [Internet]. 2020 [Consultado 8 abril 2020] Disponible en: https://www.uptodate.com/contents/genetics-clinical-features-and-diagnosis-of-marfan-syndrome-and-related
10. Sánchez Martínez R. Enfermedad de Marfan: revisión clínico terapéutica y guías de seguimiento. Seminarios de la Fundación Española de Reumatología. [Internet] 2011 [Consultado 8 abril de 2020] ;12(4):112-122. Disponible en: https://www.elsevier.es/es-revista-seminarios-fundacion-espanola-reumatologia-274-articulo-enfermedad-marfan-revision-clinicoterapeutica-guias-S1577356611000832
11. Report of the National Heart, Lung, and Blood Institute and National Marfan oundation Working Group on research in Marfan syndrome and related disorders. Circulation, 118 (2018), pp. 785-791. [Consultado 28 febrero de 2020] Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2909440/ [ Links ]

