










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Científica Ciencia Médica
versión impresa ISSN 2077-3323
Rev Cient Cienc Méd vol.22 no.1 Cochabamba 2019
CASO CLÍNICO
FENOTIPIFICACIÓN DE UN CASO DE ANEMIA DE CÉLULAS FALCIFORMES
ELUCIDATING THE PHENOTYPE IN A SICKLE CELL DISEASE CASE
Manuel Cristóbal Garcés Mendoza1,a, Daniel Alejandro Escobar Paredes2,b.
1Facultad de Ciencias de la Salud, Universidad Libre Seccional Cali. Cali, Colombia
aEstudiante de octavo semestre de medicina
bEstudiante de décimo semestre de medicina
Correspondencia a: Manuel Cristóbal Garcés Mendoza
Celular: +57 314 7748501
Correo: brigame@outlook.com
Procedencia y arbitraje: no comisionado, sometido a arbitraje externo.
Recibido para publicación: 19 de abril 2019
Aceptado para publicación: 31 de abril de 2019
Citar como: Rev Cient Cienc Med 2019; 22(1): 68-72
RESUMEN
La anemia de células falciformes es una enfermedad genética frecuente en la que la herencia de dos genes mutantes de la hemoglobina, uno de cada progenitor, produce un trastorno de la hemoglobina. Es una enfermedad crónica con exacerbaciones agudas que causan efectos a largo plazo en la educación, la vida familiar, la integración social y la calidad de vida del paciente. La clínica se resume en vaso oclusión e isquemia tisular, anemia hemolítica y la susceptibilidad a infecciones. Se presenta el caso de un hombre de raza negra, de 28 años cursando con crisis dolorosa vaso-oclusiva, trastorno hemolítico y síndrome anémico de volúmenes normales.
Palabras clave: Anemia de células falciformes; anemia hemolítica; hidroxiurea; hemoglobina fetal
ABSTRACT
Sickle cell anemia is a common genetic disease in which the inheritance of two mutant hemoglobin genes, one from each parent produces a hemoglobinopathy. It is a chronic disease with acute exacerbations that cause long-term effects on education, family life, social integration and the patient's quality of life. The clinic is summarized in vaso-occlusion and tissue ischemia, hemolytic anemia and susceptibility to infections. We present the case of a 28 years old black male, suffering from a vaso-occlusive crisis, hemolytic disorder and normocytic anemic syndrome.
Keywords: Anemia, Sickle cell ; Anemia, Hemolytic; hydroxyurea; Fetal Hemoglobin.
INTRODUCCIÓN
La anemia de células falciformes o drepanocitosis es una de las hemoglobinopatías estructurales más comunes en el mundo. Es un defecto de herencia autosómica recesiva caracterizado por la presencia de Hemoglobina S (Hb S) en el eritrocito producto de un cambio en el codón GAG normal, que pasa a GTG, lo que resulta en la sustitución del aminoácido ácido glutámico por valina, en la posición 6 de la cadena beta1.
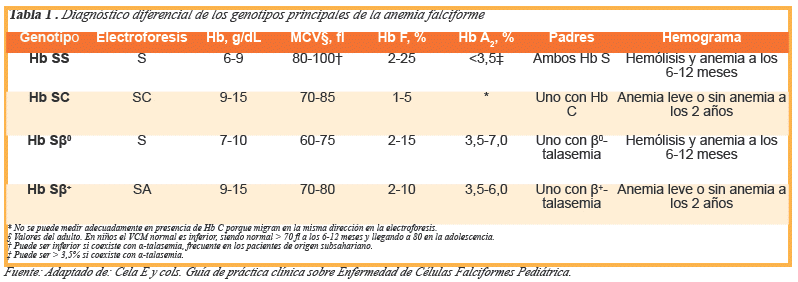
Los heterocigotos, con rasgo drepanocítico (Hb AS), son portadores asintomáticos. Los enfermos pueden ser homocigotos o dobles heterocigotos, cuando el gen anormal de la Hb S se une a otro gen anormal que afecta a la Cadena de β-globina. Entre éstos, las formas más frecuentes son la Hb SC y la Hb S-βtalasemia2, (ver Tabla 1)3.
La prevalencia del rasgo drepanocítico oscila entre 10 y 40% en el África ecuatorial y en descendientes afro americanos se presenta hasta en un 8% de la población. La enfermedad es pues característica, pero no exclusiva, de la raza negra. Existen núcleos mediterráneos con el gen de Hb S (Grecia, Italia, Turquía y norte de África) además de Arabia Saudí y la India4. En Colombia, donde el 35% de la población es afrodescendiente, hay estudios en poblaciones del Caribe y el Pacífico con hallazgos de prevalencia del rasgo falciforme de 4,5% y 10% respectivamente5,6.
La Hb S se caracteriza por polimerizarse con la desoxigenación, lo cual favorece su depósito en la membrana plasmática del eritrocito. El depósito de Hemoglobina (Hb) desnaturalizada sobre la membrana del glóbulo rojo tiene una serie de implicaciones fisiopatológicas producidas fundamentalmente por dos hechos: la distorsión del glóbulo rojo y la disfunción de las bombas de sodio.
El primer evento aumenta la rigidez de los glóbulos rojos y les confiere la forma de hoz o media luna característica de la enfermedad, estos eritrocitos rígidos se destacan por ser más lábiles que los normales (vida media menor) y menos deformables en la microcirculación, esto último provoca una seria de eventos que terminan en oclusión vascular, isquemia tisular y daño de órganos. Cuando la Hb desnaturalizada afecta las bombas iónicas de la membrana, compromete el movimiento de cationes del eritrocito lo que produce deshidratacion celular y aumenta la adherencia del hematíe al endotelio y la viscosidad sanguínea7. Por otro lado, en el bazo, el exceso de hematíes dañados sobrepasa su capacidad de filtro, impidiendo su función inmunológica (asplenia funcional) y posteriormente se produce una fibrosis progresiva (autoesplenectomía), por lo que se incrementa la susceptibilidad a infecciones8. Por tanto, la vaso oclusión e isquemia tisular, la anemia hemolítica y la susceptibilidad a infecciones son los tres problemas que condicionan la clínica de la enfermedad.
Al nacimiento los pacientes son asintomáticos por la persistencia de la Hb fetal, las primeras manifestaciones clínicas aparecen entre los 4 y 6 meses de vida cuando sus niveles disminuyen. Los rasgos clínicos característicos del paciente con anemia de células falciformes son: palidez cutánea, ictericia que se acentúa con los episodios infecciosos o vasoodusivos por aumento de la hemólisis, cambios esqueléticos por expansión de la médula ósea con deformidades a nivel de la cara y cráneo (hipertrofia de maxilares con mal oclusión dental), peso al nacimiento normal con posterior retardo marcado en el crecimiento y desarrollo, hepato y esplenomegalia en los primeros años de vida porque con la edad el bazo sufre fibrosis progresiva produciéndose auto esplenectomía en etapas posteriores, el paciente homocigoto presenta anemia crónica con aumento del gasto y el trabajo cardiaco, cardiomegalia y disminución de la tolerancia al ejercicio8-10.
Las características hematológicas de la enfermedad son anemia regenerativa, volumen corpuscular medio normal o disminuido, por otro lado, en los datos de laboratorio se encuentra evidencia de hemólisis como aumento de la bilirrubina total a expensas de la indirecta y aumento de la deshidrogenasa láctica. El diagnóstico de anemia de células falciformes se realiza sobre la base de datos clínicos o antecedentes que orienten a pensar en la presencia de drepanocitosis, acompañados de las características de laboratorio anteriormente mencionados. Se debe considerar realizar un extendido de sangre periférica en donde se van a encontrar alteraciones en la morfología del glóbulo rojo con células densas (pequeñas y deshidratadas) y en formas de hoz o disco3,10, (ver Fig. 1). Para el diagnóstico también es de utilidad la prueba de ciclaje con metabisulfito de sodio mediante la cual se evidencia la presencia de glóbulos rojos en forma de media luna (drepanocitos). La prueba confirmatoria es la electroforesis o separación cromatográfica de Hb en pH alcalino11,13. La Tabla 1 muestra los datos analíticos de cada uno de los genotipos. También están disponibles actualmente la cromatografía líquida de alta resolución y el análisis del ADN14,15.

PRESENTACIÓN DEL CASO
Paciente masculino de 28 años de edad, afro descendiente, manifiesta antecedente de anemia de células falciformes diagnosticada cuando tenía 8 meses de edad y es hospitalizado debido a un cuadro respiratorio agudo con anemia asociada, desde ese primer contacto se inició tratamiento conhidroxiurea y en su evolución requirió múltiples hospitalizaciones y transfusiones, también declara haber suspendido hace tres meses el tratamiento con hidroxiurea por problemas sociales, con antecedente paterno de la misma enfermedad, sin más antecedentes relevantes. En el momento sin estudios que confirmen el diagnóstico de anemia drepanocítica.
El paciente consulta por un cuadro clínico de dos horas de evolución consistente en dolor localizado en tórax, dorso, brazos y pelvis, constante, de intensidad 8/10. Al momento del ingreso presenta signos vitales: Frecuencia Cardiaca: 64 latidos por minuto, Frecuencia Respiratoria: 20 respiraciones por minuto, Tensión Arterial: 120/70mmHg, Temperatura: 36 °C, Saturación de O2: 97%. Paciente en buenas condiciones generales, quien se transporta por medios propios, álgido, con palidez muco cutánea, conjuntivas ictéricas, al examen físico no se encuentran más anormalidades.
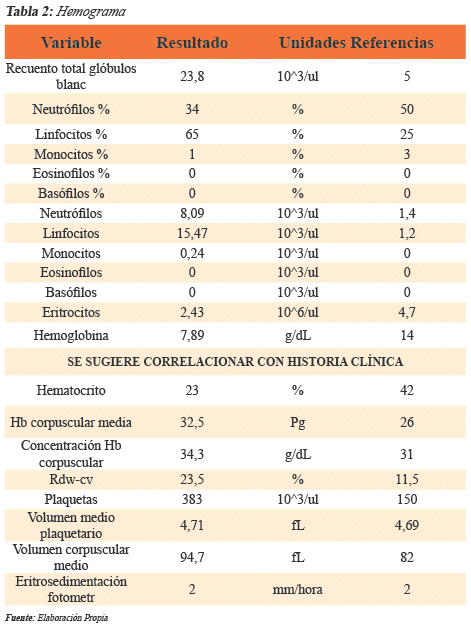
Inicialmente se le ordenan paraclínicos los cuales reportaron: un hemograma con anemia moderada de volúmenes normales (Hb 7,89 g/dl) y leucocitosis desviada a la derecha (leucocitos 23800, neutrófilos 34%, linfocitos 65%, monocitos 1%), valores elevados de deshidrogenasa láctica (1475 U/1); hiperbilirrubinemia a expensas de la bilirrubina indirecta (bilirrubina directa 0,91, bilirrubina indirecta 1,87) y radiografía de tórax sin alteraciones.
Se considera que el paciente cursaba con crisis dolorosa, síndrome anémico de volúmenes moderados y anemia hemolítica. Se inicia terapia hídrica con solución salina normal a 150 ml por hora, terapia transfusional con 2 unidades de glóbulos rojos y oxigenoterapia. Adicionalmente manejo del dolor con dipirona 1g/2ml ampolla, 2 ampollas vía endovenosa cada 6 horas, tramadol 100mg/2ml ampolla, 1 ml vía subcutánea cada 8 horas; morfina 10mg/ml ampolla, 2 ampollas vía subcutánea cada 6 horas y manejo de la patología de base con ácido fólico 1 mg tableta, 1 tableta vía oral cada 2 días e hidroxiurea 500 mg tableta, 3 tabletas cada 24 horas.
Para el diagnóstico se contó con un hemograma en el cual se evidenció anemia moderada de volúmenes normales, un aumento de la bilirrubina a expensas de la indirecta (que se puede observar tanto en hemolisis extravascular como en la hemolisis intravascular) y valores muy elevados de lactato deshidrogenasa (lo cual es sugestivo de hemólisis intravascular) (Tabla 2). En razón de que el antecedente patológico de la anemia de células falciformes no era del todo claro, ya que el paciente no tenía en su posesión alguna prueba de laboratorio que confirmara la enfermedad, se ordena un estudio electroforético de hemoglobina en medio alcalino cuyo resultado puede observarse en la "Gráfica 1". Con dicho estudio pudo evidenciarse la presencia de Hb S (62,9%) y se pudo confirmar el diagnóstico.
Con intención de vigilar la evolución del paciente se toman diariamente paraclínicos de cuadro hemático, niveles séricos de lactato deshidrogenasa y bilirrubina. El nivel de hemoglobina aumenta y los valores de bilirrubina y lactato deshidrogenasa sérica descienden progresivamente. Debido a que presenta mejoría tanto sintomática como en paraclínicos, se decide dar egreso.
DISCUSIÓN
El tratamiento de la crisis dolorosa vasooclusiva se basa en el reposo, la hidratación, oxigenoterapia y la administración de analgésicos16,17. Se tenía un paciente en el contexto clínico de una anemia moderada de volúmenes normales, evidencia de hemolisis y el antecedente parental de enfermedad de células falciformes; lo primero que se hizo fue la reposición hídrica con solución salina normal.
A continuación, evaluamos la presencia de signos de dificultad respiratoria, medimos la saturación de oxígeno e iniciamos oxigenoterapia. Existe evidencia de que las transfusiones de unidades de glóbulos rojos pueden mejorar la sintomatología del paciente que presenta anemia de moderada a severa y clínica de crisis drepanocítica, si bien no es la indicación más frecuente de transfusión en estos pacientes4, el paciente presenta al ingreso valores de hemoglobina de 7,89 g/dl y sintomatología sugestiva de crisis dolorosa por lo cual se decidió transfundir dos unidades de glóbulos rojos leucorreducidos, dado que en los siguientes días el paciente presentó mejoría del cuadro clínico, aumento de hemoglobina y disminución de marcadores de hemolisis, se decidió no realizar transfusiones adicionales. El manejo de dolor del paciente se llevó a cabo con dipirona, tramadol y morfina; la literatura recomienda administrar analgésicos a necesidad y respuesta del paciente, iniciando con al menos un opiáceo como la morfina intravenosa a 0,1 - 0,2 mg/ kg cada 4 horas o en intervalos cortos en perfusión a 0,025 mg/kg/hora. Se puede añadir ibuprofeno 5-10 mg/kg o ketorolaco 0,5 mg/kg intravenoso cada 8 horas, si el paciente es mayor de 12 años; el tramadol y la dipirona son otras alternativas17. Se reanudó tratamiento abandonado de hidroxiurea, la literatura soporta el uso de hidroxiurea como buena opción de tratamiento preventivo, ya que esta aumenta la producción de hemoglobina fetal inhibiendo la síntesis de ADN, lo que disminuye la aparición de complicaciones agudas y crónicas11. Se ha demostrado el beneficio de la administración de ácido fólico durante las anemias hemolíticas regenerativas, la dosis recomendada es de 1 mg/día12, dicha medida también se instauró en el paciente.
Se realizó electroforesis de hemoglobina en medio alcalino para establecer el diagnóstico definitivo y esclarecer la causa del cuadro clínico. La anemia de células falciformes es un síndrome que puede ser producido por más de un genotipo, algunos síndromes drepanocíticos son homocigotos, otros se producen por la herencia de Hb S de un progenitor y de otra hemoglobinopatía como la talasemia β o Hb C (doble heterocigotos), resulta invariable que un gen anormal de Hb S se una a otro gen con una mutación que también afecte a la cadena de β globina18,19. La electroforesis reporto las siguientes concentraciones de las variantes de hemoglobina: Hb A 26,5%, Hb F o variante de Hb 7,1%, Hb S 62,9%, Hb A2 3,5%. Lo cual evidencia la presencia del componente S de la hemoglobina y la combinación de un patrón electroforético AS. Ahora si bien no se trata de una anemia microcítica, los niveles de Hb A2 dentro del rango y la presencia de Hb A constituyendo el 26,5% de la Hb total, son características que permiten diagnosticar la forma Hb S-β talasemia+ que produce cierta cantidad de Hb A en el gen que se encuentra afectado de β talasemia. Con el paso de los días, el paciente mantiene una evolución hacia la mejoría tanto en síntomas como en paraclínicos por lo cual se le da egreso.
REFERENCIAS
1. Acuña C, Cuero K, Espitia K, Rojas R, Torres R. Anemia drepanocitica y situacion en Colombia: revisión. Biociencias. 2017; 3:65-75. Acceso 3 de marzo de 2018. Disponible en: http://hemeroteca.unad.edu.co/index.php/Biociencias/article/view/2242/2406 [ Links ]
2. Cervera Á, Cela E., Anemia falciforme,manejo en atencion primaria. Acceso 18 de enero de 2018. Disponible en: http://www.redalyc.org/pdf/3666/366638698009.pdf
3. Cela E, Cervera Á, Díaz CH, Rives S, Salinas JA, Sevilla J, et al. GUÍA DE PRÁCTICA CLÍNICASOBRE ENFERMEDAD DE CÉLULAS FALCIFORMES PEDIÁTRICA. Madrid: Sociedad Española de Hematología y Oncología Pediátrica; 2010. Acceso 26 de enero de 2018. Disponible en: http://www.madrid.org/cs/Satellite?blobcol=urldata&blobheader=application%2Fpdf&blobheadername1=Content-disposition&blobheadername2=cadena&blobheadervalue1=filename%3DDREPANOCITOSIS_SEHOP_2010_pdf.pdf&blobheadervalue2=language%3Des%26site%3DHospitalGregorioMaranon&blobkey=id&blobtable=MungoBlobs&blobwhere=1352892074232&ssbinary=true
4. Rendón DS, Victoria M, Jurado D. ACTUALIZACIÓN SOBRE ANEMIA DE CÉLULAS FALCIFORMES EN NIÑOS. Revista Gastrohnup. 2013; 15(2):112-8. Acceso 19 de enero de 2018. Disponible en: http://revgastrohnup.univalle.edu.co/a13v15n2/a13v15n2art8.pdf [ Links ]
5. Alvear C, Barboza M, Viola M, Monieriz C, Araque L. PILOT STUDY OF HEMOGLOBINOPAHIES IN NEWBORNS OF THE RAFAEL CALVO MATERNITY CLINIC OF CARTAGENA, COLOMBIA. Colombia Médica. 2012; 43(3):196-9. Acceso 19 de enero de 2018. Disponible en: http://colombiamedica.univalle.edu.co/index.php/comedica/article/view/925/2249 [ Links ]
6. de Bernal M, Collazos A, Bonilla RD, Tascón EP. DETERMINATION OF PREVALENCE OF HEMOGLOBIN S, C, D, and G IN NEONATES FROM BUENAVENTURA, COLOMBIA. Colombia Médica. 2010; 41(2):141-7. Acceso 19 de enero de 2018. Disponible en: http://www.redalyc.org/articulo.oa?id=28316817005 [ Links ]
7. Campos MC, Fortún A, Fortún Campo A, Hernández J. FISIOPATOLOGÍA DE LA VASO-OCLUSIÓN DE LA DREPANOCITOSIS. Rev Ciencias Médicas. 2009; 13(1):80-9. Acceso 19 de enero de 2018. Disponible en: http://www.revcmpinar.sld.cu/index.php/publicaciones/article/view/468/983 [ Links ]
8. Quintero M, Jiménez A. ANEMIA DE CÉLULAS FALCIFORMES. Revista Gastrohnup. 2012; 11(2)Supl1:S27-35. Acceso 22 de enero de 2018. Disponible en: http://revgastrohnup.univalle.edu.co/a12v14n2s1/a12v14n2s1art4.pdf [ Links ]
9. Ware RE, de Montalembert M, Tshilolo L, Abboud M. SIKLE CELL DISEASE. Lancet. 2017; 390(10091):311-23. DOI: 10.1016/S0140-6736(17)30193-9
10. Chaves W, Amador D, Sánchez J. ANEMIA DE CÉLULAS FALCIFORMES. Repert med cir. 2014; 23(3):221-5. Acceso 22 de enero de 2018. Disponible en: https://www.fucsalud.edu.co/sites/default/files/2017-01/ANEMIA%20DE%20CE%CC%81LULAS%20FALCIFORMES.pdf [ Links ]
11. Pule G, Mowla S, Novitzky N, Wiysonge C, Wonkam A. A SYSTEMATIC REVIEW OF KNOWN MECHANISMS OF HYDROXYUREA-INDUCED FETAL HAEMOGLOBIN FOR TREATMENT OF SICKLE CELL DISEASE. Expert Rev Hematol. 2015; 8(5):669–79. DOI: 10.1586/17474086.2015.1078235
12. Barraza J, Guzmán A, Vargas R. EFICACIA DE LA HIDROXIUREA EN LA DISMINUCIÓN DE CRISIS DOLOROSAS EN PACIENTES CON ANÉMIA DE CÉLULAS FALCIFORMES. BARRANQUILLA 2011-2013. Biociencias. 2014; 9(1):45-51. Acceso 26 de enero de 2018. Disponible en: http://revistas.unilibre.edu.co/index.php/biociencias/article/view/2839/2256 [ Links ]
13. Bello A. Anemia de células falciformes: Guía de manejo pediátrico. Arch Venez Puer Ped. 2012; 75 (2): 52-8. Acceso 26 de enero de 2018. Disponible en: http://www.scielo.org.ve/pdf/avpp/v75n2/art05.pdf [ Links ]
14. Fernández-García A, Álvarez-Guerra ED. LA ANEMIA DE HEMATÍES FALCIFORMES: INVESTIGACIONES PARA EL DIAGNÓSTICO Y TRATAMIENTO. Ciencia en su PC. 2007; (4)1-11. Acceso 26 de enero de 2018. Disponible en: http://www.redalyc.org/articulo.oa?id=181320170002 [ Links ]
15. Steinberg MH: Drepanocitosis y otras hemoglobinopatias. En: Goldman L, Schafer AI, eds. Goldman-Cecil Tratado de Medicina Interna. 25th Ed. Barcelona: Elsevier; 2016. p. 1095-1104
16. Simon E, Long B, Koyfman A. Emergency Medicine Management of Sickle Cell Disease Complications: An Evidence-Based Update. J Emerg Med. 2016; 51(4):370-81. DOI: 10.1016/j.jemermed.2016.05.042
17. Rojas E, Calderón E, Vidal MA, Arroyo F, García R, Tores LM. Crisis drepanocítica y tratamiento del dolor. Rev Soc Esp Dolor. 2015; 22(4):165-7. Acceso 27 de enero de 2018. Disponible en: http://scielo.isciii.es/pdf/dolor/v22n4/04_nota.pdf [ Links ]
18. Hebbel RP, Vercelloti GM: Pathobiology of sickle cell disease. En: Hoffman R, Benz E, Silberstein L, Heslop H, Weitz J, Anastasi J, eds. Hematology: Basic Principles and Practice. 7th ed. Philadelphia: Elsevier; 2018. p. 571-583.
19. Martín MR, Frómeta ED, Domínguez MM, Díaz KA, Santana HM, Viñales PM. Anemia Falciforme: Conocimientos y percepción actual del riesgo en jóvenes detectados al nacimiento como portadores sanos. RevistaCubana de Genética Comunitaria. 2008; 2(3).Acceso 26 de enero de 2018. Disponible en: http://bvs.sld.cu/revistas/rcgc/v2n3/rcgc090308.htm [ Links ]
20. Torrens M. Interpretación clínica del hemograma. Rev. Med. Clin. Condes. 2015; 26(6):713-25. Acceso 1 de marzo de 2018. Disponible en: http://www.elsevier.es/index.php?p=revista&pRevista=pdf-simple&pii=S0716864015001480&r=202 [ Links ]

