










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Científica Ciencia Médica
versión impresa ISSN 2077-3323
Rev Cient Cienc Méd vol.20 no.2 Cochabamba 2017
Caso Clínico
PRESENTACIÓN DE UN CASO: SÍNDROME DE CLAUDE BERNARD-HORNER POST RESECCIÓN DE HIGROMA QUÍSTICO.
PRESENTATION OF A CASE: CLAUDE BERNARD-HORNER'S SYNDROME POST RESECTION OF CYSTIC HYGROMA.
Samuel Andrés Barrientos1, Mitchell Senaedy Borjas1, Karla Alejandra Lagos1, Dr. Douglas Varela2.
1 Carrera de Medicina, Facultad de Ciencias Médicas de la Universidad Nacional Autónoma de Honduras.
2 Asesor del caso clínico.
Correspondencia a: Karla Alejandra Lagos Correo electrónico: karlita_lagos@hotmail.com
Procedencia y arbitraje: no comisionado, sometido a arbitraje externo.
Recibido para publicación: 13 de octubre 2016
Aceptado para publicación: 15 de noviembre 2017
Citar como: Rev Cient Cienc Med 2017; 20 (2): 57-61.
RESUMEN
El Higroma quístico es una malformación congénita que consiste en uno o más espacios linfáticos llenos de líquido, suelen reconocerse en recién nacidos, pero típicamente pueden ser visibles cuando aumentan de tamaño conforme al crecimiento del paciente. El Síndrome de Claude Bernard-Horner se caracteriza por una miosis, ptosis palpebral y anhidrosis en pacientes con afectación de la vía oculosimpatica, se considera una complicación neurológica iatrogénica infrecuente.
Se presenta una paciente femenina de 4 años con diagnostico posnatal de higroma quístico cervical tras estudio patológico realizado, el cual fue extraído quirúrgicamente. Tres días después la paciente presenta ptosis del parpado superior izquierdo con presencia de miosis y disminución de la sudoración del mismo lado, sin alteraciones en la fuerza y movimiento del miembro superior Izquierdo.
Palabras clave: higroma quístico, ptosis palpebral, miosis, anhidrosis.
ABSTRACT
Cystic hygroma is a congenital malformation consisting of one or more lymphatic spaces filled with fluid, usually recognized in newborns, but typically can be visible when they increase in size according to the patient's growth. The Claude Bernard-Horner Syndrome is characterized by miosis, palpebral ptosis and anhidrosis in patients with oculosympathetic pathway involvement, it is considered an infrequent iatrogenic neurological complication. We present a 4-year-old female patient with postnatal diagnosis of cervical cystic hygroma after a pathological study, which was surgically removed. Three days later, the patient presented ptosis of the left upper eyelid with the presence of miosis and decreased sweating on the same side, without alterations in the strength and movement of the left upper limb.
Keywords: cystic hygroma, eyelid ptosis, miosis, anhidrosis.
INTRODUCCIÓN
En 1869, el oftalmólogo suizo Johann Friedrich Horner descubrió una paciente de 40 años presentándose con cefalea, ptosis palpebral, miosis y eritema facial derecho, lo cual atribuyó a un daño de la vía oculosimpática. Anteriormente el estudio publicado en 1852 por el fisiólogo francés Claude Bernard describió el mismo cuadro en animales con lesiones de vías nerviosas. Por tanto, la asociación de miosis, enoftalmos y ptosis palpebral homolateral con compromiso de la cadena simpática cervical se conoce como Síndrome de Claude Bernard-Horner1,2.
Se conoce como Higroma Quístico o Linfangioma Quístico a la presencia de tumoraciones benignas únicas o múltiples de características quísticas que ocurren predominantemente en la infancia, a menudo se presentan en el área de la cabeza y el cuello que afecta a los vasos linfáticos yugulares e impide el su correcto drenaje, son una anomalía congénita sin distinción entre sexos y con un diagnóstico principalmente clínico3,4,5,6.
PRESENTACION DEL CASO
Se presenta a una paciente de 4 años del sexo femenino con antecedente de masa blanda (8 x 8 cm aprox.) presente desde los 15 días de vida, localizada en región laterocervical izquierda, ubicada desde el área mastoidea hacia el área supraclavicular; depresible, no supurativa. Tres días posteriores a la consulta medica manifiesta presencia de calor, dolor a la palpación y fiebre subjetivamente alta. Al examen físico se resalta la presencia de amigdalas hipertróficas e hiperemicas sin exudados visibles.
De manera preliminar se diagnostico como un quiste braquial por el servicio de pediatría, posteriormente, luego de una revaloracion, fue cambiado por el diagnostico de higroma quístico. Al presentar cambios inflamatorios se considero un proceso infeccioso en la paciente, motivo por el cual se inicio cobertura antibiotica (Ceftriaxone) acompañado de antiiflamatorio no esteoideo (Diclofenac) pese a la existencia de valores normales dentro del hemograma, la quimica sanguinea y una temperatura de 37°C el mismo dia. Asimismo se solicitó estudios de imagen para ampliar el abordaje de la patologia y cuatro días después se cancela el plan de cobertura farmacológica.
El resultado de la Tomografia Axial Computarizada (TAC) reportó una masa con bordes bien definidos, densidad líquida con afectación de los diferentes niveles cervicales izquierdos, con medidas de 9,5x7cm y volumen de 97cc. La masa produce una importante compresión de estructuras adyacentes incluyendo la via aérea, solicitándose una pronta extirpación quirúrgica. (ver figura A)
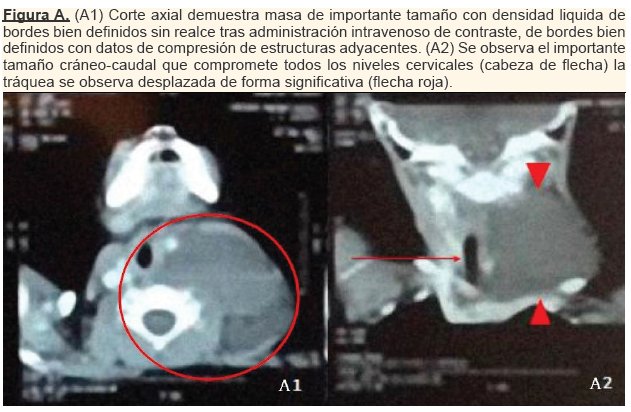
Hallazgo de cirugía: Masa cervical quística encapsulada de aproximadamente de 15 cm, irregular, vascularizada, con desplazamiento traqueal y con adherencia a vena yugular/arteria carótida; presenta contenido seroso, sangrado hemático de 120 ml. Se realizó una biopsia para su estudio en el departamento de patología.
En el postoperatorio la paciente presentó esfuerzo respiratorio motivo por el cual se acopló a ventilador mecánico, asimismo presentó leve anisocoria no reactiva a la luz con midriasis en ojo derecho (OD) más que en ojo izquierdo (OI). Luego de la intervención quirúrgica y de la anestesia respectiva, la paciente permaneció dormida y tardó 6 horas en recuperarse en su totalidad.
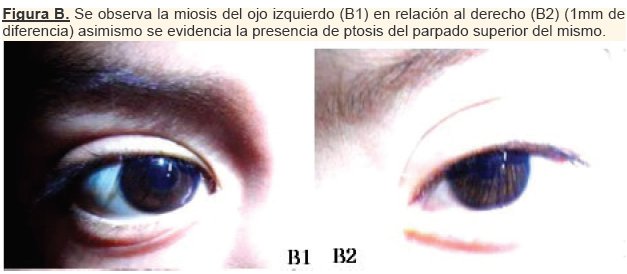
En el segundo día post-operatorio el cuello se encontraba simétrico, con una herida quirúrgica limpia, sin cambios inflamatorios, no habia existencia de enfisema subcutáneo. Al examen físico se encontró anisocoria, siendo el diametro de la pupila izquierda inferior a la pupila derecha; asimismo se observó la presencia de ptosis palpebral del ojo izquierdo.
En el tercer dia post-operatorio se remitió a neurología pediátrica por presentar ptosis palpebral de (1-2mm) del ojo izquierdo con diametro pupilar de 3mm en OD y 2mm en OI, sin enoftalmos, acompañado de anhidrosis del mismo lado, diagnosticándose de esta manera el Síndrome de Claude Bernard Horner. (ver figura B y C)

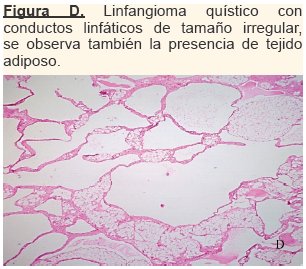
Hallazgo de biopsia,- Descripción Morfológica: Lamina de fragmento ovalado de tejido que mide 9.5x7x3.5cm con áreas quísticas de contenido cetrino y zonas semiblandas; al corte se evidencian espacios quísticos con aspecto en panal de abeja y con contenido amarillo verdoso junto a múltiples espacios quísticos uniloculares de diferentes tamaños. El diagnóstico histopatológico corresponde a un Higroma Quístico. (ver figura D)
DISCUSIÓN
El Síndrome de Claude Bernard-Horner, en su forma completa se aprecia anhidrosis facial ipsilateral y puede ser acompañado por heterocromía del iris en caso de lesiones congénitas. Suele ser congénita (que está presente desde el nacimiento), aunque también existe una forma adquirida cuya causa se asocia con frecuencia a simpatectomías (interrupción quirúrgica de parte del de las vías nerviosas simpáticas , realizada para aliviar el dolor crónico) y otras cirugías cervicales en adultos7.
Anatomía: La vía simpática tiene tres niveles o divisiones (no se decusa en ninguno de ellos). La vía Central (neurona de primer orden), nace en el hipotálamo posterolateral y desciende dorsolateralmente por el tronco encefálico hasta el centro cilio espinal de Budge-Waller en la sustancia gris de la médula a nivel de C8-T2, seguidamente la vía preganglionar (neurona de segundo orden) cruza el ápex pulmonar, pasa por ganglio estrellado, cruza bajo la arteria subclavia y asciende por el cuello para hacer sinapsis en el ganglio cervical superior y finalmente la vía postganglionar (neurona de tercer orden) es donde las fibras simpáticas de la cara y del ojo se separan. Si la lesión es preganglionar y la vía postganglionar está intacta se produce la dilatación de la pupila afectada en el 90% de los casos8. En las lesiones preganglionares la anhidrosis y su distribución comprometen el cráneo, cara, cuello y bajo la clavícula. Las lesiones post ganglionares pueden ser extra o intracraneanas1,8.
Clínica: Ptosis: La pérdida de la inervación simpática de los músculos del tarso superior e inferior condiciona ptosis unilateral que nunca es completa1. Miosis: Se produce por parálisis del músculo dilatador del iris. La Anisocoria generalmente es pequeña (< 1mm), es mayor en la oscuridad y puede desaparecer en la luz por acción del esfínter de la pupila (parasimpático) sin presencia de síntomas visuales generalmente1 . Anhidrosis: Las fibras simpáticas sudomotoras de la cara después de la sinapsis en el ganglio cervical superior, llevan solo unas pocas de estas fibras a la piel de la frente. Por esto la anhidrosis solo ocurre en pacientes con afectación central o preganglionar1. En forma aguda y debido a la pérdida del control vasomotor del lado afectado, se produce aumento de la temperatura acompañado de eritema unilateral facial, epifora, hiperemia conjuntival y secreción nasal. Posteriormente, por desarrollo de hipersensibilidad simpática, se produce vasoconstricción, con disminución de la temperatura del lado afectado, palidez y disminución de la sudoración8,9.
El Higroma Quístico se encuentra en un número dispar dentro de los desórdenes genéticos y no genéticos, su incidencia es variable aunque globalmente es de aproximadamente un caso por cada 50,000 recién nacidos vivos6,10. Un higroma quístico ocurre a medida que el bebé crece en el útero, este se desarrolla a partir del saco endolinfático primitivo embriónico yugular. Generalmente luce como una protuberancia suave bajo la piel, típicamente crece junto con el crecimiento del niño y cerca de 90% de los casos se hace evidente después de los dos años de edad3,9,11. Dentro de los tumores cervicales fetales, el linfagioma o higroma quístico es el más frecuente11, el 75% de estas neoplasias se originan en la región cervical, mientras que el resto puede localizarse en diversas áreas como la axila, región inguinal o en el mediastino5. Clínicamente estos tumores se presentan como una masa cervical quística única o múltiple de tamaño variable, consistencia blanda, indolora, mal delimitada, fluctuante, lobulada, multitabicada, translúcida, contienen un líquido claro y transparente, no adherida a tejidos profundos y la piel que lo cubre puede ser delgada. Puede presentar exacerbaciones secundarias a infecciones, hemorragia o sin explicación aparente que puede causar una insuficiencia respiratoria por comprensión de la vía aérea. Su crecimiento es lento y progresivo a lo largo del primer año de vida y puede producir trastornos circulatorios por compresión3,6,9. El diagnóstico es fundamentalmente clínico, la transiluminación orienta hacia el contenido líquido del tumor. La ecografía cervical, radiografía de tórax y tomografía axial computarizada ayudan a evaluar la extensión y características. El tratamiento consiste en la extirpación completa del tejido anormal, sin embargo los higromas quísticos con frecuencia pueden crecer, lo que hace imposible la extirpación de todo el tejido. El pronóstico es bueno si se puede extirpar totalmente el tejido anormal con cirugía3,9. Las complicaciones más frecuentes son las infecciones sobre agregadas en etapa postnatal, hemorragia, trombosis sobreañadida con fenómenos organizativos y de recanalización de los trombos. Los cambios morfológicos también van a depender del órgano o tejido donde asiente, ya que en estructuras laxas el crecimiento será mayor. Otra complicación que puede abarcar es el daño a las estructuras del cuello causados por la cirugía, así como la reaparición del higroma cuando no se logra extirpar en su totalidad3,5,6,9.
El Síndrome de Claude Bernard-Horner debido a procesos a nivel cervical de pacientes es una afectación bastante infrecuente11 ya que este puede ser el resultado a una multitud de procesos con diferente pronostico12, ya sea por cirugías de cuello, anestésicos locales, congénito, compresión local por un hematoma o un tumor, incluso como consecuencia de un inadecuado posicionamiento peri operatorio, siendo estos procesos en los cuales el síndrome se establece como resultado de la parálisis de la cadena simpática cervical ipsilateral tras una lesión del ganglio estrellado13,15. El Higroma quístico se presenta como una masa cervical quística única o múltiple, de crecimiento lento y progresivo a lo largo del primer año de vida con un diagnóstico principalmente clínico. El tratamiento consiste en la resección quirúrgica y su incidencia global aproximada es de 1/50,000 recién nacidos vivos8,10. Según un estudio dentro de la población pediátrica correspondiente en el hospital de Bagdad, las masas en el cuello de los pacientes pediátricos son distribuidas con un patrón y distribución similar a nivel mundial, mostrándose una variación únicamente en la categoría de infecciones e inflamaciones vinculadas a estas, ya que dependen del estado socioeconómico del paciente, patógenos según área y otros factores14. Los linfagiomas cervicales suelen llamar la atención al existir algún proceso infeccioso por la inflamación que estos llegan a presentar, pero pueden llegar a ser una patología grave tanto por compresión de la vía aérea, al obstruir estructuras vitales como arterias y venas en cuello o por sangrados masivos a los cuales son predisponentes por su morfología. La compresión de estructuras nerviosas se encuentra como un evento a considerar también, aunque con una incidencia menor en relación a las anteriores. Siendo el tratamiento del Higroma de orden quirúrgico, en el cual la remoción total del tejido tiene un valor fundamental, se cuenta con la probabilidad de que existan lesiones a nivel de estructuras vasculares, respiratorias y nerviosas. Es aquí donde la experiencia y efectividad del cirujano influye notablemente en este proceso. En cambio, en los reportes donde se discuten entre las diferentes secuelas a largo plazo tras un proceso quirúrgico a nivel cervical, se han descrito previamente en adultos la existencia del síndrome de Claude Bernard-Horner, tras una resección de linfagioma quístico cervical16.
Exponemos así que los higromas quísticos de cuello pese a ser malformaciones linfáticas17, mediante la extirpación quirúrgica del mismo como tratamiento de elección en la actualidad, puede conllevar a la existencia del Síndrome de Claude Bernard - Horner como consecuencia iatrogénica en infantes de misma manera que en adultos.
REFERENCIAS
1. Fariñas Z, Rangel R, Guerra R. Síndrome de Claude-Bernard-Horner. Presentación de un caso. Acta Medica del Centro. 2012. 6(4): 96. Citado el 5 de junio 2015. Disponible en: http://www.medigraphic.com/pdfs/medicadelcentro/mec-2012/mec124o.pdf [ Links ]
2. Dogliotti A. Conceptos actuales en la parálisis braquial perinatal: Parte 1: etapa temprana. Arch. argent. pediatr. 2011. (4): 347-353. Citado el 22 de abril de 2017. Disponible en: http://www.scielo.org.ar/scielo.php?pid=S0325-00752011000400011&script=sci_arttext&tlng=pt [ Links ]
3. Barriga J, Murillo C, Agreda J. Higroma quístico, a propósito de un caso. Revista de la Sociedad Boliviana de Pediatría. 2002; 41(2). Citado el 22 de abril del 2017. Disponible en: http://www.scielo.org.bo/scielo.php?pid=S1024-06752002000200010&script=sci_arttext [ Links ]
4. Brunori EA, Caratozzolo G, Martinez M, Dinerstein NA. Higroma Quístico a Propósito de un Caso. Revista Hospital Materno Infantil Ramón Sarda. 1996; XV (2). Acceso 18 junio 2015. Disponible en: http://www.sarda.org.ar/Profesionales/Publicaciones/Revista_Sarda/1996/HIGROMA_QUISTICO_A_PROPOSITO_DE_UN_CASO [ Links ]
5. Huera SI., Elias J. Linfangioma cervical fetal: Diagnóstico prenatal y resultado perinatal. Revista Peruana de Ginecología y Obstetricia. 2012. Acceso 19 junio 2015. Disponible en: http://www.scielo.org.pe/scielo.php?pid=S2304-51322012000400013&script=sci_arttext [ Links ]
6. Síndrome de Horner secundario a cirugía cervical. Acta Otorrinolaringológica Española. 2012; 63(4): 299-302. Citado el 22 April del 2017. Disponible en: http://linkinghub.elsevier.com/retrieve/pii/S0001651912000635?via=sd [ Links ]
7. Síndrome de Horner secundario a anestesia epidural. 2007; 22(3):196-200. Citado el 22 abril del 2017. Disponible en: http://neurorgs.net/wp-content/uploads/Investigacion/neuroanestesia/sindrome-horner-secundario-anestesia-epidural.pdf
8. Hospital Privado de Comunidad. El síndrome de Claude Bernard-Horner y otros desconocidos de siempre. 5(2) Acceso el 12 junio del 2015. Disponible en: http://www.hpc.org.ar/v2/v_art_rev.asp?id=15&offset=11
9. Sumana M. Cystic hygroma: MedlinePlus Medical Encyclopedia. Salud.wikiplus.org. 2015. Citado el 22 April 2017. Disponible en: http://salud.wikiplus.org/medlineplus/ency/article/000148.htm
10. Aranguez M., Palmer P, Gonzales I, Illero J.M. Aspectos clínicos y morfológicos de los linfangiomas infantiles: Revisión de 145 casos 1996. Citado el 22 Abril del 2017. Disponible en: https://www.aeped.es/sites/default/files/anales/45-1-6.
11. Galeana Castillo C, Casas Patiño D, Rodríguez Torres A. Correlación de la imagen ecográfica y patológica de higroma quístico.Archivos de Investigación Materno Infantil.2013; V (2). Acceso el 18 junio 2015. Disponible en: http://www.medigraphic.com/pdfs/imi/imi-2013/imi132h.pdf. [ Links ]
12. Garcia R, Fernandez M, Russinyol J. Higroma Quístico del Cuello.Revista Cubana de Pediatría. 2006; 78(2). Acceso el 18 junio del 2015. Disponible en: http://scielo.sld.cu/pdf/ped/v78n2/ped12206.pdf [ Links ]
13. Cavidad Puentes A, Bermudez Guerrero F, Zea Medina M.V. Síndrome de Horner contralateral asociado a bloqueo de ganglio estrellado: reporte de caso y revisión de la literatura.Revista de la Sociedad Española del Dolor. 2014; 21(4). Acceso el 18 junio del 2015. Disponible en: http://scielo.isciii.es/pdf/dolor/v21n4/07_revision.pdf [ Links ]
14. Mayoof AF. Neck masses in paediatric population: An experience with children attended the Central Teaching Hospital of Pediatrics in Baghdad 2008-2009. Afr J Paediatr Surg. 2015; 2(12):136-9. Disponible en: http://www.afrjpaedsurg.org/text.asp?2015/12/2/136/160364 [ Links ]
15. Vicente P, Canelles E, Díaz A, Fons A. Síndrome de Horner irreversible tras simpatectomía toracoscópica bilateral. Archivos de la Sociedad Española de Oftalmología. 2014; 89(2):79-81. Citado el 2 de julio del 2015. Disponible en: http://linkinghub.elsevier.com/retrieve/pii/S0365669112004042?via=sd [ Links ]
16. Molino J.A, Guillen G., García-Vaquero J.A, Peiro J, Marhuenda C, Carreras E. Linfangioma quístico cervical: Todavía un reto.Revista de Cirugía Pediátrica.2010; 23(3). Acceso el 1 julio del 2015. Disponible en: http://www.secipe.org/coldata/upload/revista/2010_23-3_147-152.pdf [ Links ]
17. Bindu P.R, Mathew J, Thomas P, Jayanyhi P, Krishnapillai R. Cavernous lymphangioma of lower lip. CASE REPORTS. 2013; 2(3). Acceso el 2 Julio del 2015. Disponible en: http://healthsciences.ac.in/jul-sep-13/6.ACasereport1.html [ Links ]

