










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Científica Ciencia Médica
versión impresa ISSN 2077-3323
Rev Cient Cienc Méd vol.18 no.2 Cochabamba 2015
REPORTE DE CASO
Dermatomiositis juvenil, una patologia infrecuente en la infancia: Reporte de un caso
Juvenile dermatomyositis, a disease uncommon in childhood: A case report
Mayra Victoria-Rocha Choque1,2; Zaida Ríos Saavedra1; Dra. Vianca Pereira3; Dr. Marcelo Luizaga3
1Facultad de Medicina "Dr. Aurelio Melean". Universidad Mayor de San Simón. Cochabamba-Bolivia
2Sociedad Científica de Estudiantes de Medicina
3Reumatólogo Pediatra Hospital Manuel Ascencio Villarroel-Universidad Federal de rio De Janeiro-Hospital Universitario Clementino Fraga Filho.
Correspondencia a: Mayra Victoria Rocha Choque
mayrarocha6892@gmail.com
Procedencia y arbitraje: no comisionado, sometido a arbitraje externo.
Recibido para publicación: 20 de octubre del 2015
Aceptado para publicación: 10 de diciembre del 2015
Citar como: Rev Cient Cienc Med 2015; 18(2): 48-52
Abreviaturas utilizadas en este artículo:
EAS = Autoinmunes sistémicas
DMJ= Dermatomiositis Juvenil
IV= Intravenosa
VO= Vía oral
PRINTO= Organización Internacional de Ensayos de Reumatología Pediátrica
RESUMEN
La Dermatomiositis Juvenil es una miopatía autoinmune de la infancia rara con una incidencia de 3 casos por cada 1 000 000 habitantes, con una relación mujer-hombre 2:1, la edad de presentación es bimodal con el primer pico a los 5 años y el segundo a los 11 años. Se manifiesta con afectación cutánea y muscular. Se presenta el caso de un paciente masculino de 6 años de edad afectado de dermatomiositis juvenil, diagnosticado en el Hospital Manuel Ascencio Villarroel por presentar debilidad muscular proximal, eritema en heliotropo con edema a nivel periorbicular, pápulas de gottron en articulación metacarpofalangicas y calcinosis en codos. Se inició tratamiento con meltilprednisona en 2 bolos, seguido de Prednisona oral. Se evidencia mejoría progresiva del paciente en siguientes visitas a consulta externa tras instalado el tratamiento. Esta patología plantea problemas diagnósticos, nosológicos y terapéuticos, generalmente tras un diagnóstico y tratamiento oportuno el pronóstico del paciente es bueno.
Palabras clave: Dermatomiositis, Poliomiositis, Miositis
ABSTRACT
Juvenile dermatomyositis is a rare autoimmune myopathy childhood with an incidence of 3 cases per 1 million inhabitants, with a female-male ratio 2:1, the age of presentation is the first bimodal peak at age 5 and the second to 11 years. It manifests with skin and muscle involvement. For a male patient affected 6-year-old juvenile dermatomyositis diagnosed in the Manuel Ascencio Villarroel Hospital with proximal muscle weakness, heliotrope erythema in conjunction with periorbicular level edema, papules Gottron metacarpoflangicas joint and calcinosis occurs in both elbows.Treatment was started with 2 bowling meltilprednisona, followed by oral prednisone. The patient shows progressive improvement once time that treatment was installed.This condition raises diagnostics, nosological and therapeutic problems, but after a diagnosis and treatment the patient's prognosis is usually good.
Keywords: Dermatomyositis, Polymyositis, Myositis
INTRODUCCIÓN
Las enfermedades autoinmunes sistémicas (EAS) actualmente desplazaron el término de colagenopatías, haciendo notar su participación orgánica más allá del daño al colágeno que antes las delimitaba1.
La Dermatomiositis Juvenil (DMJ) es una miopatía autoinmune de la infancia que pertenece al grupo de las EAS, en la instalación del cuadro se presenta vasculitis de pequeños vasos de gravedad variable y de forma tardía la calcinosis2,3. Es una patología infrecuente, registrándose 3 casos por cada 1 000 000 habitantes, con una relación mujer-hombre 2:1, la edad de presentación es bimodal, con un primer pico entre los 5 a 9 años y entre los 11 a 14 años4,5.
La fisiopatología de esta entidad se basa en la destrucción de fibras musculares y afectación cutánea ligada a infiltrado inflamatorio. El mecanismo inicial se desconoce, se cree que existe un factor desencadenante como una infección, toxico, o fármaco, que produce una activación de células endoteliales y la sobreexpresión por las fibras musculares estriadas de antígenos de histo-compatibilidad de clase I, conjuntamente con una predisposición genética, lo que lleva a la activación del sistema inmunitario con una citotoxicidad celular y humoral que produce a una destrucción muscular 6,7.
El diagnóstico clínico se basa en debilidad muscular proximal simétrica, manifestaciones cutáneas y hallazgos anormales enzimáticos, electromiográficos e histológicos3. La terapia con corticoide asociados a inmunosupresores ha demostrado ser eficiente6. El seguimiento del tratamiento médico debe llevarse a cabo con rehabilitación muscular, control metabólico y evaluaciones periódicas5.
Debido a la incidencia infrecuente de esta patología en Bolivia, además de la dificultad diagnóstica que representa esta entidad por manifestación tardía de lesiones cutáneas, presentamos a continuación un caso de Dermatomiositis Juvenil y una revisión bibliográfica.
PRESENTACIÓN DEL CASO
Paciente masculino de 6 años de edad, procedente de Cochabamba Cercado, acude a consulta externa del Hospital Manuel Ascencio Villarroel por presentar calcinosis en articulación del codo, rodillas y región retro auricular acompañado de debilidad muscular de extremidades superiores e inferiores.
Fue diagnosticado previamente de Distrofia Muscular de Duchenne hace 2 años, por presentar debilidad muscular proximal simétrica progresiva de inicio en miembros inferiores con elevación marcada de las enzimas musculares. En los últimos 9 meses la debilidad muscular proximal simétrica se acentuó en cintura pélvica y escapular, hasta dificultar la deambulación, con compromiso muscular de tronco y cuello, lo que dificulta la capacidad de levantar la cabeza y levantarse de la cama, además de presentar artromialgias discretas simétricas en muñecas y articulaciones interfalangicas proximales bilaterales. No refiere antecedentes patológicos y hereditarios relevantes.
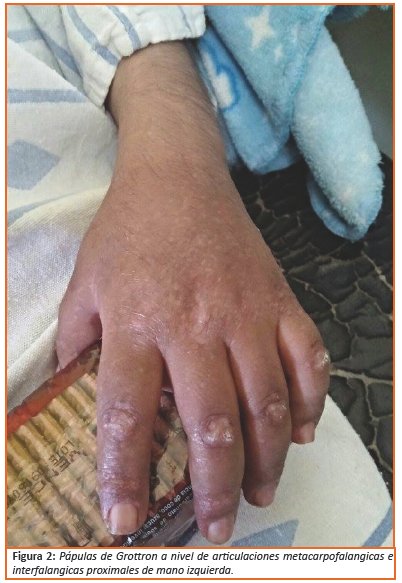
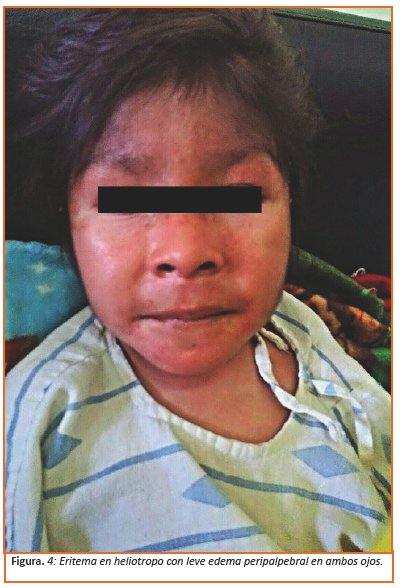
Al examen físico niño de 15 kg de peso, 99 cm de talla, con 36.6 °C de temperatura, frecuencia respiratoria de 26 rpm, frecuencia cardiaca de 98 lpm y presión arterial 85/60 mmHg, en regular estado general en decúbito dorsal, consciente, normohidratado, hemodinámicamente estable. Se evidencian lesiones cutáneas: calcinosis circunscritas en ambos pabellones auriculares, codo (Ver Figura 1), rodillas dolorosas a la presión, lesiones eritematosas en cara, frente, mejillas, pirámide nasal (eritema en heliotropo), ligero edema en región facial, más acentuado en región peripalpebral (Ver Figura 2); pápulas de gottron en articulación metacarpo falángicas e interfalángicas proximales de forma bilateral (Ver Figura 3), además de presentar hipertricosis generalizada. También presentaba lesiones descamativas en cuero cabelludo y lesiones hipercrómicas en piel de tórax anterior y posterior. Miembros inferiores y superiores con debilidad muscular proximal simétrica con disminución de la fuerza muscular 1/5 llevando a la incapacidad funcional, reflejos osteotendinosos disminuidos en miembros superiores y ausentes en miembros inferiores.
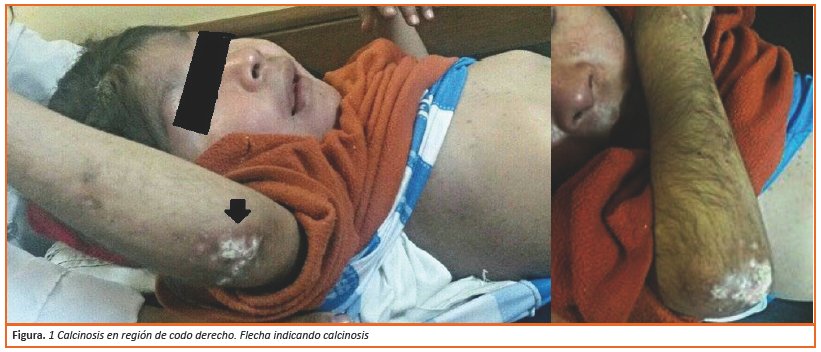
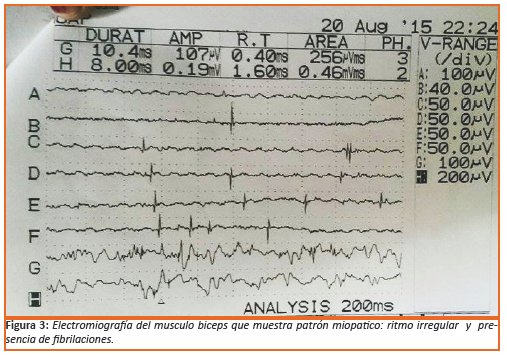
Los exámenes complementarios solicitados fueron radiografía de tórax, manos y codos sin alteraciones. Los laboratorios de ingreso mostraron: GOT 80 UI, GPT 54 UI, proteínas totales 9,2 g., globulina 5,3 g., Fosfatasa Alcalina 259 UI, LDH 529 UI, CPK - MB 516 UI. Se solicitó una electromiografía el cual fue compatible con cambios de tipo miopático, mediante la identificación de tres características: fibrilaciones, potenciales de unidad motora disminuida y disminución de la amplitud de onda (Ver Figura 4). Se solicitó una biopsia muscular del cual aún no se recibieron los resultados.
Las interconsultas se realizaron con el servicio de Dermatología y Reumatología pediátrica, los cuales evaluaron las características clínicas cutáneas como neuromusculares respectivamente y en asociación con exámenes complementarios se cumplen con los criterios diagnósticos para DMJ y se inicia tratamiento.
Entre los diagnósticos diferenciales que fueron tomados en cuenta figuran entidades con compromiso neuromuscular: Distrofia Muscular de Duchenne y Polimiositis. La distrofia fue considerada por el antecedente de diagnóstico en otro complejo hospitalario por la debilidad muscular que usualmente comienza en piernas y pelvis, dificultad de habilidades motoras de evolución progresiva, el diagnóstico de Polimiositis fue considerado por la debilidad muscular simétrica proximal de ambos miembros superiores e inferiores, sin duda las lesiones cutáneas principalmente la calcinosis oriento al diagnóstico definitivo, ya que sin la presencia de esta ultima el cuadro solo se hubiera diagnosticado como polimiositis.
Se inició tratamiento con bolos de Metilprednisolona 400 mg por vía intravenosa (IV) y 24 hrs después se inicia con Prednisona 3-4 mg/kg/día por vía oral (VO) asociado a Metotrexate 10 mg/ semana, para las lesiones dermatológicas se indicó ácido fusidico ungüento al 2%, crema de urea se le indico 2 veces al día sobre la lesión cutánea.
En próxima consulta de aproximadamente un mes dado de alta, se evidencia mejoría clínica significativa, con el tratamiento de Prednisona oral y los ungüentos para las lesiones dermatológicas, ya no se observa calcinosis en piel y la fuerza muscular mejoro a 2/5, con predominio en cintura pélvica. Tras la mejoría clínica se indica iniciar Fisioterapia de Rehabilitación.
DISCUSIÓN
La DMJ es una entidad patológica rara que para su diagnóstico necesita de criterios puntuales que se basan desde la evidencia clínica hasta la confirmación mediante exámenes complementarios, pero no se evidencia una asociación de estos desde fases iniciales de la enfermedad en la mayor parte de los casos5,6.
Las alteraciones cutáneas y musculares solo se encuentran afectadas de forma simultánea en un 60% de los casos, y el solo 30% presenta manifestaciones cutáneas que preceden a las musculares5. En el caso de este paciente la sintomatología muscular precedió a las lesiones cutáneas, esto puede llevar a confusión en el diagnostico con otras patologías como Distrofia muscular de Duchenne.
Ante la variabilidad de manifestaciones clínicas. En 1975, Bohan y Peter agrupa estos criterios en las siguientes categorías: Diagnostico sospechoso, probable y definitivo7.
Estos criterios diagnósticos incluyen los siguientes parámetros: debilidad muscular proximal que puede ir acompañado o no de disfagia y afectación de los músculos respiratorios, aumento de las enzimas musculares, afectación cutánea típica: exantema en heliotropo sobre los párpados y pápulas de Gottron a nivel de las articulaciones metacarpofalángicas e interfalángicas (y en otras articulaciones como tobillos, rodillas, codos), biopsia muscular compatible con miopatía, y el estudio de electromiografía : potenciales de unidades motoras cortos, polifásicos, fibrilaciones2,6.
El presente caso pertenece a la categoría de DMJ definitivo por la presentación de 4 de los 5 criterios diagnósticos, cabe recalcar que las características dermatológicas orientaron al diagnóstico definitivo6,7.
Respecto al tratamiento, este debe ser individualizado la evolución de la enfermedad el cual va desde un cuadro clínico leve de debilidad muscular y manifestaciones cutáneas hasta un cuadro de gravedad con debilidad muscular generalizada que llega a comprometer la musculatura respiratoria y que puede llevar a la muerte del paciente, el tratamiento además incluye el control de la miositis inflamatoria subyacente, la prevención y / o tratamiento de las complicaciones7,8. Si bien existen pocos datos de ensayos controlados aleatorizados para guiar el tratamiento en los niños con DMJ, actualmente se utilizan varios esquemas alternativos basados en estudios observacionales y de experiencia clínica7,9.
El tratamiento inicial es una combinación de glucocorticoides asociado a un inmunosupresor: dosis altas de prednisona oral (2 mg/kg por día) y metotrexate (15 mg/m2 una vez por semana,) por vía oral o subcutánea. A falta de respuesta a la terapia se añade otro inmunosupresor asociado como la ciclosporina (3 a 5 mg/kg por día), ajustando la dosis del medicamento dependiendo de la respuesta clínica. Los niños que reciben metotrexate también deben recibir ácido fólico (1 mg por día) para limitar su toxicidad, excepto el día de uso del metotrexate3,10.
La Organización Internacional de Ensayos de Reumatología Pediátrica (PRINTO) ha llevado a cabo un ensayo multicéntrico que comparó tres regímenes de tratamiento (prednisona sola, prednisona más metotrexate y prednisona en combinación con ciclosporina). Los datos preliminares sugieren que ya sea metotrexato o ciclosporina pueden actuar como agentes ahorradores de esteroides con la misma eficacia, pero hay menos efectos adversos con metotrexate10.
Se indica el uso de metilprednisolona IV (pulsos) en lugar de prednisona oral en las siguientes situaciones: pacientes con enfermedad ulcerosa o una mala respuesta o empeoramiento de la enfermedad con terapia oral, personas con compromiso respiratorio que no pueden tomar medicamentos orales, y los que cursan con complicaciones gastrointestinales que pueden limitar la absorción, se debe considerar todas estas situaciones para adaptar al paciente a un tratamiento efectivo9,10.
Se reserva el uso de ciclofosfamida en pacientes con enfermedad grave, enfermedad ulcerosa crónica del tracto gastrointestinal, piel, o problemas respiratorios y en pacientes con DMJ recurrentes o con falla terapéutica a los medicamentos antes mencionados, se opta por el uso de la inmunoglobulina intravenosa (IGIV) (dosis de 2 a 4 g /kg divido en dos o cinco días)3,10.
En el caso de nuestro paciente se inició solamente con un pulso de metilprednisolona por ser un caso crónico de más de un año de evolución y por las características clínicas, no se llegó a realizar tres pulsos como protocolo de tratamiento debido a falta de recursos económicos, además que el paciente se encontraba con desnutrición, por lo que se optó por continuar el tratamiento con corticoide vía oral a dosis máximas.
Respecto al pronóstico de los pacientes con DMJ, este resulta ser bastante variable. En un extremo están los pacientes que logran remisiones definitivas con terapia glucocorticoide y en el otro extremo están los que no responden a ninguna terapia que inevitablemente desarrollan atrofia muscular con severa impotencia funcional e incluso la muerte por afectación de otros sistemas6. Entre estos dos extremos se encuentran la mayoría de los pacientes que presentan recaídas y remisiones6. Pero actualmente el pronóstico en los pacientes afectados con esta entidad mejoró considerablemente desde la introducción de la terapia inmunosupresora teniendo una disminución de la tasa de mortalidad, actualmente la mortalidad global estimada es de 15%, pero se evidencia que aún existe morbilidad, esto debido a que la calcinosis cutánea circunscrita grave y extensa puede causar mayor discapacidad a largo plazo pudiendo presentar dolor crónico, ulceración persistente asociada a infección, formación de abscesos y contracturas articulares2,11.
Entre los factores de mal pronóstico se encuentran diagnóstico tardío y retardo en el inicio de tratamiento, fibrosis pulmonar, compromiso miocárdico y disfagia4. El compromiso pulmonar intersticial se asocia a debilidad de músculos respiratorios e infecciones que facilita el desarrollo de neumonías aspirativas4,6, La disfagia debe considerarse como marcador de gravedad de las miopatías inflamatorias ya que se ha asociado a pacientes con mayor compromiso muscular y sistémico6.
Dentro la literatura se encontró los siguientes reportes de caso: primero una paciente femenina de 4 años y 5 meses con diagnóstico previo de lupus eritematoso sistémico y con desarrollo posterior de manifestaciones clínicas cutáneas y musculares de DMJ en los siguientes dos años sin presencia de complicaciones, completó un tratamiento con bolos de metilprednisolona con un total de 20 dosis cursando posteriormente con mejoría importante, con regresión notable de la calcinosis, eritema facial leve y fuerza muscular 4/5, persiste leve contractura articular por desuso2; otra paciente de sexo femenino de 12 años con DMJ, con antecedente de dos episodios de neumonía en los últimos 6 meses, a su ingreso presentaba clínica muscular y cutánea propia de DMJ, se le indico el tratamiento con prednisona oral con metotrexate por 4 semanas curso con mejoría clínica inicial de lesiones cutáneas pero persistiendo debilidad muscular en cintura escapular, reingreso al complejo hospitalario por presentar disfagia leve por lo cual se le inicio bolos de metilprednisolona por tres días, evolucionó favorablemente, con desaparición de la disfagia y mejoría de la debilidad muscular4; otro fue un paciente masculino de 2 años con un cuadro de evolución de tres semanas de debilidad muscular, en los últimos 15 días presento manifestaciones cutáneas curso con tratamiento con tres bolos de metilprednisolona con posterior inicio de prednisona oral durante un año, a los seis meses posteriores el paciente recuperó totalmente la funcionalidad muscular y remisión de lesiones cutáneas12.
Comparando el presente caso con otros reportes, concluimos que nuestro paciente si bien se encontraba en un estadio avanzado con un buen tiempo de instalación de las manifestaciones musculares, cabe resaltar que no presentaba complicaciones sistémicas ni ningún otro factor de mal pronóstico que comprometa su vida, por lo cual tras iniciado el tratamiento y posterior evidencia de la mejoría clínica progresiva, nuestro paciente presenta un pronóstico favorable.
Agradecimientos sinceros a nuestro docente Dr. Carlos Erostegui Revilla por su colaboración en la redacción, revisión del trabajo y apoyo en la interpretación del estudio de electromiografía.
REFERENCIAS
1. Cairoli E. Unidades de enfermedades autoinmunes sistémicas: notas de una experiencia en curso. Rev Méd. Urug 2013; (29):248-9. Acceso en: Agosto 2015. Disponible en: http://www.scielo.edu.uy/scielo.php?pid=S1688-03902013000400008&script=sci_arttext
2. Morel Z, Martinez R, Mendieta S, et al. Dermatomiositis juvenil y calcinosis extensa. Tratamiento con metilprednisolona y metotrexato. Reumatol Clin 2008; 4(6):248-50. Acceso en: Agosto 2015. Disponible en: http://www.reumatologiaclinica.org/es/dermatomiositis-juvenil-calcinosis-extensa-tratamiento/articulo/S1699258X08755463/
3. Maldonado C, Martinez V, Navarrete G, Ríos H. Dermatomiositis idiopática primaria. Dermatol. Rev Mex 2012; 56(5):308-17. Acceso en: Agosto 2015. Disponible en: http://www.medigraphic.com/pdfs/derrevmex/rmd-2012/rmd125e.pdf
4. Sánchez M, Climent H, Fonseca L, et al. Dermatomiositis. Aportación de un caso de afectación leve-moderada y disfagia precoz. An Pediatr (Barc) 2015; 82(1):86-89. Acceso en: Agosto 2015. Disponible en: http://www.sciencedirect.com/science/article/pii/S1695403313004426
5. Aldana N, Casale V, Olmos J. Dermatomiositis: presentación de un caso y reseña bibliográfica. Salud(i) Ciencia 2015; (21): 317-20. Acceso en: Agosto 2015. Disponible en: https://www.siicsalud.com/dato/sic/213/141003.pdf
6. Lioger B, Lavigne C, Machet L. Dermatomiositis. EMC – Dermatología 2011;(45):1-12. Acceso en: Agosto 2015. Disponible en: http://www.sciencedirect.com/science/article/pii/S1761289611710433
7. Selva A.M, Trallero E. Miopatías inflamatorias. Dermatomiositis, polimiositis y miositis con cuerpos de inclusión. Reumatol Clin 2008; 4(5):197-206. Acceso en: Agosto 2015. Disponible en: http://www.sciencedirect.com/science/article/pii/S1699258X08724641
8. Allenbach Y, Benveniste O. Poliomiositis, dermatomiositis y otras miopatias inflamatorias idiopáticas. EMC 2015;4 8(1):1-11. Acceso en: Agosto 2015. Disponible en: http://www.em-consulte.com/es/article/958065/polimiositis-dermatomiositis-y-otras-miopatias-inf
9. Yanez J, Cisternas M, Saldias V, Saldias F. Dermatomiositis refractaria asociada a neumonía en organización tratada con rituximab. Reporte de un caso. Rev Méd Chile 2009; 137: 88-93. Acceso en: Agosto 2015. Disponible en: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0034-98872009000100013
10. Hutchinson C Feldman B, Thomas J, Patterson M. El tratamiento y el pronóstico de la dermatomiositis juvenil y polimiositis. UptoDate 2015 Acceso en: Agosto 2015. Disponible en: http://www.uptodate.com/contents/treatment-and-prognosis-of-juvenile-dermatomyositis-and-polymyositis?source=search_result&search=youth+dermatomyositis&selectedTitle=3~144
11. Ciudad C, Avilés JA, Campos M, et al. Dermatomiositis: estudio y seguimiento de 20 pacientes. Actas Dermosifiliogr 2011; 102(6):448-55. Acceso en: Agosto 2015. Disponible en: http://apps.elsevier.es/watermark/ctl_servlet?_f=10&pident_articulo=90021222&pident_usuario=0&pcontactid=&pident_revista=103&ty=153&accion=L&origen=zonadelectura&web=www.elsevier.es&lan=es&fichero=103v102n06a90021222pdf001.pdf
12. Mainou A, Mainou C, Plaza F, et al. Dermatomiositis juvenile d’inici precoç. Pediatr Catalana 2010; 70(1):158-61 Acceso en: Agosto 2015. Disponible en: http://dialnet.unirioja.es/servlet/articulo?codigo=3582737

