










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Científica Ciencia Médica
versión impresa ISSN 2077-3323
Rev Cient Cienc Méd vol.18 no.1 Cochabamba 2015
ARTÍCULO DE REVISIÓN
Trombofilias hereditarias
Hereditary Thrombophilia
Carlos Danilo Noroña Calvachi 1,2
1 Médico General
2 Médico Adjunto, Centro de Biomedicina, Universidad Central del Ecuador
Correspondencia a: Carlos Danilo Noroña Calvachi
Correo electrónico: carlosdnoronac@gmail.com
Procedencia y arbitraje: no comisionado, sometido a arbitraje externo.
Recibido para publicación: 30 de Agosto de 2014
Aceptado para publicación: 15 de Enero de 2015
Citar como: Rev Cient Cienc Med 2015; 18(1): 43-49
Abreviaturas utilizadas en este Artículo
TEV = La tromboembolia venosa
GRADE = Grading of Recommendations Assessment, Development and Evaluation (Clasificación de las Recomendaciones de Evaluación, Desarrollo y Evaluación)
TP = Tiempo de protrombina.
TTP = Tiempo de tromboplastina
TTPa = Tiempo de tromboplastina activada.
TT = Tiempo de trombina.
INR = International Normalized Ratio
ELISA = Enzyme-Linked Immuno Sorbent Assay(Ensayo por inmunoabsorción ligado a enzimas)
RESUMEN
Las trombofilias hereditarias suponen un grupo de enfermedades que predisponen al desarrollo de enfermedad tromboembólica arterial y venosa, debido a déficit o ganancia de función de factores anticoagulantes o procoagulantes incrementando de manera significativa la morbilidad y mortalidad en la población adulta y pediátrica. La expresión y penetrancia genética de este grupo de enfermedades es diversa, y las formas de presentación clínica varía desde la purpura fulminans neonatal hasta episodios tromboembólicos recurrentes a edades tempranas y efectos adversos en el embarazo. El screening no es rutinario en pacientes con cuadros tromboembólicos y sus indicaciones son precisas, en especial personas menores a los 45 años, con cuadros recurrentes, y abortos o muertes fetales a repetición sin causa específica. El tratamiento es basado de acuerdo a la presentación del cuadro clínico, sin embargo la anticoagulación convencional es ampliamente utilizada en el manejo de este grupo de pacientes.
Palabras clave: Trombofilia, Factor V Leiden, Déficit de Proteína C, Tromboembolismo Venoso, Anticoagulante
ABSTRACT
Inherited thrombophilia represent a group of diseases that predispose to the development of arterial and venous thromboembolic disease due to deficiency or gain of function of anticoagulant or procoagulant factors, increasing significantly the morbidity and mortality in the adult and pediatric population. Expression and genetic penetrance to this group of diseases is diverse, and the form of clinical presentation varies from the neonatal purple fulminans to recurrent thromboembolic events at a young age and pregnancy with side effects. The screening is not routine in patients with thromboembolic condition and its indications are accurate, especially in younger people than 45, with recurrent episodes of abortions or fetal deaths without specific cause. Treatment is based according to the clinical presentation of the condition; however conventional anticoagulation is widely used in the treatment of this patient group.
Keywords: Thrombophilia, Factor V Leiden, Protein C Deficiency, Venous Thromboembolism, Anticoagulant
INTRODUCCIÓN
A la trombofilia se la ha definido como un desorden de la coagulación que aumenta la predisposición a la formación de coágulos incrementando de forma significativa la morbilidad y mortalidad descrita a inicios de los años 60 cuando se presentaron trombosis venosas profundas con amplio componente familiar, manifestándose como causa al menos 250 mutaciones de la Antitrombina, trombofilia descrita por primera vez. La tromboembolia venosa (TEV) se halla fuertemente aso ciada a la presencia de una o más condiciones de trombofilia, la cual puede ser adquirida o hereditaria. La presencia del Factor V Leiden y la mutación del gen de la protrombina (F2G20210A) son las afecciones más comunes, y es asociada con aumento del riesgo de desarrollo de trombosis venosa profunda y tromboembolia pulmonar entre el 5 a 80%, este último dato respecto al estado homocigoto en el Factor V Leiden, mientras que la deficiencia de Proteína C, Proteína S y Antitrombina III el riesgo llega hasta el 20%, siendo la homocigosis de la Proteína S la que peor resultado clínico presenta, por tanto la presencia de heterocigosis u homocigosis determina tanto el resultado clínico como la variabilidad de la presentación1.
Las manifestaciones clínicas son ampliamente variadas y están muy relacionadas con la edad de presentación, factores de riesgo asociados, estado de heterocigoto u homocigoto de la enfermedad que finalmente dan lugar a cuadros de trombosis venosa y en algunos trombosis arterial como cuadro inicial, en muchas ocasiones en territorios "no usuales" como el seno venoso sagital, venas mesentéricas, vena portal, vena renal, entre otras, que pueden ser manejados apropiadamente con anticoagulación convencional, sin embargo la presencia de trombofilia, puede determinar riesgo de recurrencia, también se presentan cuadros potencialmente letales como el caso de la purpura fulminans neonatal y purpura fulminans con coagulación intravascular diseminada en especial neonatos con deficiencia de Proteína C1,2.
El tratamiento se basa en anticoagulación convencional, en relación a las guías internacionales se extiende desde 3 a 6 meses y consejería genética en caso de embarazo. El screening no está rutinariamente indicado, pero si en circunstancias como edad de trombosis menor a los 50 años, historia familiar y personal, pérdidas recurrentes del embarazo2.
METODOLOGÍA DE BÚSQUEDA DE ARTÍCULOS, NIVELES DE EVIDENCIA
Se realizó búsqueda en la base de datos PubMed, EMBASE, Medline, y MD Consult (Enero 2006 a Agosto 2014) de artículos en el idioma inglés, y en Scielo (Enero 2006 a Diciembre del 2013), utilizando los términos MeSH: "inherited thrombophilia" or "hypercoagulable states" in combination with "diagnosis", "epidemiology", "review", "laboratory test", "patho-physiology", "pregnancy", "children" "management", "deep venous thrombosis", y en español con los términos "trombofilia", "déficit de proteína C y S", "anticoagulación", se priorizaron los estudios, reportes de caso y revisiones de los últimos cinco años, sin embargo, se ha incluido artículos desde el 2006 en la lista final de referencias.
La calidad de evidencia y niveles de recomendación se los ha calificado de acuerdo al sistema GRADE (Grading of Recommendations Assessment, Development and Evaluation) Modificado (Guyatt, et al. 2006), donde las recomendaciones se las realiza de acuerdo al siguiente sistema: 1A (recomendación fuerte con evidencia de calidad alta), 1B (recomendación fuerte, con evidencia de calidad moderada), 1C (recomendación fuerte, con calidad de evidenciabaja o muy baja), 2A (recomendación débil, con calidad de evidencia alta), 2B (recomendación débil, con calidad de evidencia moderada) y 2C (recomendación débil, con calidad de evidencia baja o muy baja) (Guyatt G, et. al.).
EPIDEMIOLOGÍA
Las trombofilias se presentan entre 2.8 a 7.9% de las trombosis venosas profundas, y de los pacientes que desarrollan fenómenos tromboembólicos venosos incluso en estados de gestación al menos el 30 al 50% presentan una trombofilia2.
Las deficiencias por pérdida de función presentan una prevalencia entre el 1-3/100000 anualmente, individualmente la deficiencia de la Antitrombina es una condición bastante rara, con una prevalencia 1/2000 anualmente, es la primera trombofilia descrita; en el caso del déficit de la Proteína C se presenta entre el 0.14 a 0.5% de la población general, el déficit de Proteína S entre el 0.01 a 1% (Tabla 1)2,3.
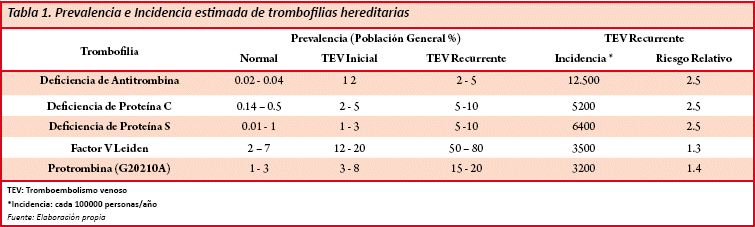
Las mutaciones que causan ganancia de función suele ser variable, la presencia del Factor V Leiden se ha demostrado en el 2 a 7% (Tabla 1) de la población general y es la mutación más común, pero es muy raro en asiáticos y africanos donde su prevalencia es menor a 1%, el estado homocigoto se encuentra en 1/2500 hasta 1/100000 en cambio el heterocigoto en el 0.1 a 0.3% de portadores de la mutación, en el embarazo se estima su presencia en un 0.26% en tromboembolias venosas asintomáticas, y de un 17% en tromboembolias venosas sintomáticas. La mutación del gen de la protrombina por su parte afecta entre 1 a 6% de la población general, en el embarazo cuando se desarrolla tromboembolias venosas sintomáticas su prevalencia es del 16.9%3,4.
COAGULACIÓN Y FISIOPATOLOGÍA DE LAS TROMBOFILIAS
La coagulación presenta una serie de reacciones proteolíticas hasta la generación de trombina, tras la presencia de una injuria vascular por medio de las vías intrínseca, extrínseca y común. Es conocido que las enzimas que participan activamente en esta cascada se hallan inactivas en forma de zimógenos y el clivaje proteico entre los diversos factores procoagulantes activa a estos catalizadores. La cascada de la coagulación se inicia una vez que existe un daño vascular lo cual desencadena la activación de Factor VII (VIIa) mediado por calcio, el cual se une con el Factor Tisular en el endotelio en el sitio de la lesión vascular formando así un complejo Factor VIIa-Factor Tisular, este complejo activa al Factor IX (IXa) y al Factor X (Xa) completando así la vía extrínseca de la coagulación; por otro lado la exposición de colágeno por parte de las células que sufren daño en la injuria vascular da lugar a la activación del Factor XII (XIIa) y este a su vez a la activación del Factor XI (XIa) actuando luego sobre el Factor IX transformándolo a Factor IX activado (IXa), finalmente llega hasta el Factor Xa donde termina la vía intrínseca de la coagulación. En la vía común de la coagulación toma gran importancia el Factor Xa, y en especial su asociación con el Factor V, el cual una vez activado (Va) formará el complejo protrombinasa que permite la conversión eficaz desde Protrombina hasta Trombina, el cual a su vez produce la activación continua del Factor V y el Factor VIII (VIIIa, el mismo además sigue activando al Factor X) (Figura 1)5,6.
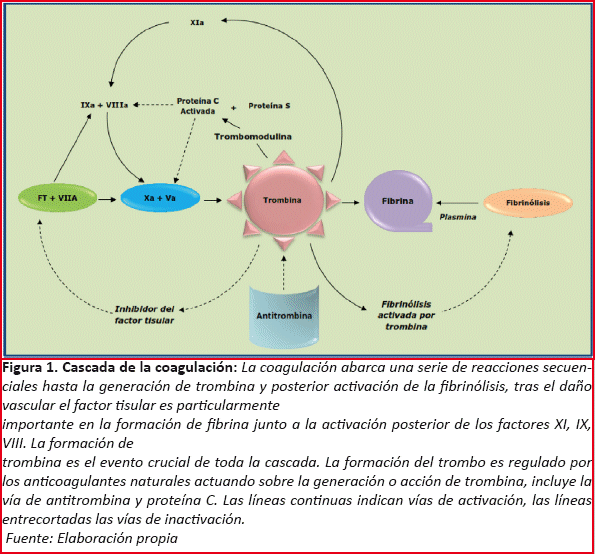
La formación de trombina da lugar a una nueva serie de eventos, ya que posee una actividad protrombótica y anticoagulante de forma indirecta, en cuanto a la primera, la trombina se une al fibrinógeno transformándola hasta fibrinógeno insoluble que forma el tapón hemostático, además de la activación de los factores V y VIII, también lo hace con los factores XI y XIII. Su actividad anticoagulante radica en la activación de la Proteína C una vez que forma parte del complejo trombina-trombomodulina a nivel endotelial; esto permite que la Proteína C activada (APC, por su siglas en inglés activated protein C) se una con su cofactor Proteína S y logre una unión efectiva en las zonas de clivaje del Factor V activado produciendo disminución de su actividad. La anticoagulación natural se completa con la Antitrombina III (ATIII), cuya función toma importancia una vez que inactiva al Factor Xa y a la trombina principalmente, aunque sus efectos también intervienen en los factores IXa, XIa, XIIa deteniendo la cascada de la coagulación, mejorando su acción si se asocia heparina(Figura 1)6.
FACTORES PROCUAGULANTES Y LA FISIOPATOLOGÍA DE LA TROMBOFILIA
Factor V: es parte del complejo protrombinasa, que permite junto al factor Xa generar trombina, en su control efectivo se da gracias a la acción de la APC que se da gracias a la unión en una de las tres zonas de clivaje que el factor presenta que son: Arg306, Arg679 y Arg506, se demuestra la importancia de la zona Arg 506 para la exposición y posterior unión de la Proteína C activada a las zonas Arg 306 y 679 asociando la mutación puntual por sustitución de arginina por glutamina en la región 506 (ahora Arg506Gln) a la falta de susceptibilidad a la inactivación por APC, y ausencia de clivaje en las zonas 306 y 679 por ausencia de exposición, lo cual conlleva a la presencia del Factor V Leiden, que mantiene activo al Factor VIII y la producción continua de trombina junto al Factor Xa6.
Factor II, Protrombina: la protrombina es activada por el complejo protrombinasa para la generación de trombina, promueve además la activación de los factores Vy VIII. Poort (1996), describe la variación de la región 3' no traducible de la secuencia que codifica el gen de la protrombina, lo que conduce a la sustitución de adenina por guanina en el nucleótido 20210, dando lugar a la mutación del gen en su expresión final (F2G20210A) que promueve un aumento por ganancia de función en la producción del factor por tanto en generación de trombina. La heterocigosis del alelo para la mutación, hace que se encuentre un 30% más cantidad de factor en el plasma6.
FACTORES ANTICOAGULANTES Y LA FISIOPATOLOGÍA DE LA TROMBOFILIA
Antitrombina III: es codificado por el gen SERPINC1 en el cromosoma 1 (1q25.1) y su mutación por pérdida, inserciones, dan lugar a una reducción en su actividad efectiva como anticoagulante, describiéndose al momento 250 mutaciones. Presenta tres tipos diversos; el tipo 1 se caracteriza por producción disminuida de antitrombina normal, el tipo 2 dado por mutaciones debido a sustitución de aminoácidos en el sitio activo y da lugar a una serinproteasa poco funcional con cantidades normales en el plasma con baja actividad asociada a heparina, y el tipo 3 se desarrolla por mutaciones en la zona de unión de la heparina, dando lugar a antitrombina con función y cantidad preservada pero con escasa función asociado a heparina, que hace que los factores Xa y trombina mantengan su función procoagulante por desregulación, y el tratamiento con heparina no sea eficaz en cuadros tromboembólicos venosos6,7.
Proteína C: es codificado por el gen PROC 1 en el cromosoma 2 (2q13.14), y se presentan mutaciones puntuales, inserciones y mutaciones por pérdida, que provoca una disminución en su función inhibitoria sobre el factor V y su capacidad de activarse tras su interacción con el complejo trombina-trombomodulina. Se describe dos tipos; el tipo 1 se relaciona a una producción de proteína normal pero a niveles extremadamente bajos, al menos 50% del valor normal, y el tipo 2 que da lugar a una proteína funcionalmente deficiente pero en cantidad normal, que provoca en primera instancia que la activación inicial sea pobre y su unión al factor V deficiente provocando estado de hipercoagulabilidad6,7,8.
Proteína S: codificada en el cromosoma 3 por el gen PROS 1, presenta 3 tipos de mutación; el tipo 1 con cantidades casi indetectables de proteína, el tipo 2 con actividad disminuida y el tipo 3 con fracción libre y actividad deficiente, no se lleva a cabo su función de cofactor, y hace que la inhibición de factor V sea débil, provocando el estado protrombótico6,7.
MANIFESTACIONES CLÍNICAS
La clínica de las trombofilias hereditarias muestra una amplia gama de manifestaciones y puede ser incluso asintomática8. Tanto en pacientes adultos y pediátricos se presenta trombosis venosas profundas a repetición comúnmente en miembros inferiores, sin embargo, una de las características de esta etiología es su distribución en sitios anatómicos poco usuales como: seno venoso sagital, miembros superiores, vena renal, venas mesentéricas, sistema portal, venas de drenaje suprarrenal, venas retinianas, con un patrón de inicio a edades tempranas sin factores de riesgo atribuibles que pueden llegar hasta el tromboembolismo pulmonar como desenlace final sin una anticoagulación adecuada9,10.
Existen manifestaciones con alto potencial de mortalidad a corto plazo como en el caso de la purpura fulminans neonatal que se caracteriza por aparición de púrpura en región de occipucio, nalgas, miembros inferiores que progresa hasta coagulación intravascular diseminada en horas o días, debido al déficit de proteína C en estado homocigoto11,12. También se ha descrito a la tormenta trombótica, que es definida por la Thrombotic Storm Study Group (2010) como una serie de eventos trombóticos agudos y subagudos con desarrollo en días o semanas, en personas jóvenes, con afecciones trombóticas arteriales y venosos en especial en sitios anatómicos poco usuales, con recurrencia inexplicable, refractaria al tratamiento anticoagulante convencional, ampliamente visto en pacientes en unidades de cuidado intensivo, asociado a trombofilias hereditarias combinadas13.
Los cuadros clínicos sutiles de las trombofilias hereditarias se ha estudiado en las manifestaciones cutáneas, que dan incluso un índice diagnóstico hacia la patología responsable, en este contexto la necrosis cutánea inducida por warfarina es una de las entidades más descritas, se observa 3 a 5 días después de iniciar warfarina y se observa en déficit de proteína C y S. El livedo reticularis caracterizado por el aspecto símil a una red vascular de color violáceo en miembros inferiores, se presenta en el déficit de proteína C, proteína S y Antitrombina III; la variante livedo vasculopático se ha asociado a la presencia del Factor V Leiden y de la mutación G20210A de la protrombina14.
En los estados de gestación, las manifestaciones son diversas y con gran repercusión en el curso del embarazo y del resultado global, casi en el 23% de pacientes con presencia de trombofilia desarrollaran tromboembolismo venoso sintomático o asintomático15. Sin embargo, las pérdidas del embarazo de forma temprana (antes de las 10 semanas) se ha asociado con los déficit de proteína S, Factor V Leiden y déficit de antitrombina III, las pérdidas tardías tras las 10 semanas se presentan en la presencia de Factor V Leiden y mutación G20210A de la protrombina. El Factor V Leiden además se lo ha relacionado con el riesgo de desarrollo de preeclampsia severa, en controversia, según Robertson et.al OR: 2.19 (IC 95%: 1.46-3.27) y al retraso de crecimiento intrauterino (RCIU), según Roque et. al OR: 12.93 (IC 95%: 2.72-61.45)16.
DIAGNÓSTICO Y SCREENING
El diagnóstico de las trombofilias hereditarias se basa en una adecuada investigación de historia personal y familiar antes de considerar un análisis específico para trombofilia. El análisis inicial en cuanto a laboratorio en este grupo de pacientes debe incluir análisis de coagulación con tiempo de protrombina (TP), tiempo de tromboplastina (TTP), tiempo de tromboplastina activada (TTPa), tiempo de trombina (TT) y fibrinógeno, así como test de función hepática, renal, biometría hemática completa y análisis de orina en busca de proteinuria17,18.
Los métodos utilizados para definir la trombofilia son: análisis de ADN para el gen de protrombina, estudio con TTPa para Factor V Leiden complementado con análisis de ADN para mutación de Factor V Leiden para confirmar, en los déficit de proteína C y S test cromogénico funcional complementado con inmuno ensayo (ELISA) para definir el tipo de déficit, que deben ser propuestos cuando la sospecha diagnóstica es alta, la cual puede ser diferida, excepto en algunos casos como la resistencia al tratamiento con heparina que amerita estudio de déficit de antitrombina III, presencia de purpura fulminans neonatal que decide estudio inmediato de proteína C y S19,20.
El screening con las técnicas moleculares para estudio de trombofilias no son indicadas de rutina, pero pueden considerarse cuando se identifican dos o más factores de riesgo, entre los que destaca edad de presentación previa a los 50 años, TVP recurrentes y en sitios inusuales, historia familiar (Tabla 2).

Algunas consideraciones antes de la realización de los exámenes son: no encontrarse en tratamiento actual con warfarina esperando al menos 4 semanas tras la finalización del tratamiento, esperar 6 meses luego del cuadro agudo de trombosis, algunos test como proteína C y S deben hacerse antes del embarazo si hay alto riesgo obstétrico, cuando no sea posible suspender la anticoagulación se puede reemplazar a warfarina por heparina de bajo peso molecular por 10 a 14 días hasta la realización del examen20,21.
TRATAMIENTO
El manejo de los cuadros trombóticos relacionados con trombofilias primarias se la realiza con anticoagulación estándar (1B) y en caso graves anticoagulación agresivas (1B), el tiempo de anticoagulación varía al contexto clínico y personal del paciente, y según la British Society for Haematology define que la anticoagulación se extienda entre 6 semanas a 3 meses, en algunos casos de por vida considerando si el episodio trombótico ocurre con factor desencadenante, factores de riesgo asociados y riesgo de sangrado asociado a la anticoagulación (1B) (Tabla 3)22.
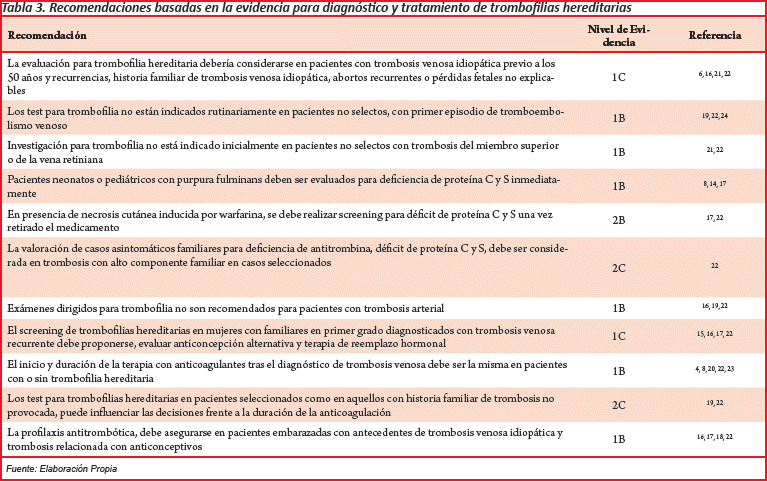
La anticoagulación se la realiza con heparina no fraccionada en infusión o con heparinas de bajo peso molecular, con o sin asociación con warfarina. En el caso de la infusión con heparina no fraccionada se debe vigilar de forma estricta el tiempo de tromboplastina (TTP), y deberá recibir alta warfarina. Si se decide el uso de warfarina se la debe realizar a dosis bajas ante el riesgo de reducción máxima de proteína C y S (riesgo de estado protrombótico), ajustando las dosis de acuerdo al International Normalized Ratio (INR), debe asegurarse la anticoagulación hasta el cuarto día tras el inicio de warfarina con heparina no fraccionada o heparina de bajo peso molecular cuando deben ser retiradas22,23,24.
La monitorización de éxito en el tratamiento con heparinas de bajo peso molecular se la realiza con cuantificación de Factor Xa que debe establecerse entre 0.5 a 1, y en el caso de warfarina con un INR entre 2 a 323,24.
En el caso de púrpura fulminans neonatal y profilaxis para procedimientos invasivos y cirugías la Guideline for the Prevention and Treatment of Venous Thromboembolism recomiendo el uso de concentrados de Proteína C con dosis inicial de 100 UI/kg, con mantenimiento de 50 UI/kg c/8-12 horas19,25.
DISCUSIÓN
Las trombofilias hereditarias tienen una baja prevalencia en los escenarios clínicos por lo que es indispensable realizar un adecuado diagnóstico diferencial para su diagnóstico y tratamiento final basado en factores de riesgo e indicaciones precisas de tamizaje8.
La identificación de factores de riesgo es una herramienta eficaz hacia la sospecha de una trombofilia hereditaria siendo el antecedente familiar de tromboembolismo venoso recurrente en miembros de primer grado, edad de inicio del primer cuadro de trombosis previo a los 50 años, y resultados adversos en el embarazo (abortos a repetición) los que presentan mayor asociación con estos trastornos y son indicaciones de screening dependiendo de la situación clínica5,20.
Los métodos de screening no son rutinarios en el caso de un tromboembolismo venoso, y tras determinar el riesgo de acuerdo a los antecedentes las indicaciones son claras, sin embargo, en los países envías de desarrollo el acceso a los test específicos de tamizaje y posterior diagnóstico significan un gran gasto y costo en salud, pues las técnicas utilizadas como ELISA, inmunoensayo para determinar funcionalidad, estudios de ADN y pruebas cromogénicas ameritan un gran recurso técnico y una interpretación adecuada para un diagnóstico final exacto que muchas veces puede resultar negativo sin que esto ayude al tratamiento y conducta final hacia el cuadro clínico, por tanto solo se los reserva como estudios de seguimiento una vez resuelto el cuadro trombótico y en situaciones que signifiquen riesgo vital como la púrpura fulminans neonatal14,17.
La identificación conjunta de trombofilias hereditarias y adquiridas se mantiene como una conducta adecuada en obstetricia, fundamentalmente a mujeres que presenten factores de alto riesgo obstétrico que según la American Journal of Obstetrics and Gynecology consisten en abortos tempranos sin causa aparente, eventos trombóticos en el embarazo, puerperio o muertes fetales de causa no determinada, de tal manera que permita un diagnóstico prenatal y reducciones de riesgo a través de tromboprofilaxis18,21.
Nuevos estudios son necesarios en cuanto a la duración de la terapia con anticoagulantes orales, pues el diagnóstico de trombofilia primaria no determina que el tratamiento se extienda de por vida, excepto en trastornos combinados o recurrencias con periodos asintomáticos cortos, también acerca de las indicaciones de tromboprofilaxis en el paciente pediátrico, en especial en ictus cerebral cuya indicación aún no es totalmente recomendada22.
CONCLUSIONES
Las trombofilias hereditarias son un grupo raro de enfermedad y aumentan el riesgo global en el desarrollo de tromboembolismo venoso y resultados adversos en el embarazo. Los pacientes con trombofilia pueden ser asintomáticos o presentar cuadros trombóticos severos en algunos casos mortales. El screening no es rutinario, pero debe investigarse cuando se encuentran factores de riesgo y cuadros clínicos críticos. El tratamiento se basa en anticoagulación convencional y prevención de las recurrencias.
REFERENCIAS
1. Burnett B. Managing of Venous Thromboembolism. Prim Care Clin Office Pract 2013; 40(1):73–90. Acceso 11 de agosto 2014. Disponible en: http://www.sciencedirect.com/science/article/pii/S0095454312001005
2. Pabinger I, et. al. Mortality and inherited thrombophilia: results from the European Prospective Cohort on Thrombophilia.
J Thromb Haemost 2012; 10(1):217–22. Acceso 8 de agosto 2014. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2011.04573.x/pdf
3. Roddis J. Managing hereditary thrombophilia. Nurs Times 2011; 107(14):15-7. Acceso 10 de agosto 2014. Disponible en: http://www.nursingtimes.net/nursing-practice/specialisms/haematology/managing-hereditary-thrombophilia/5028409.article [ Links ]
4. Rahman A, Islam M, Husnayen S. Recurrent Deep Vein Thrombosis due to Thrombophilia. Korean Circ J 2012; 42(5):345-48. Acceso 11 de agosto 2014. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3369967/pdf/kcj-42-345.pdf [ Links ]
5. Kahn S, Dickerman J. Hereditary thrombophilia. Thromb J 2006; 4(15):1-15. Acceso 31 de julio 2014. Disponible en: http://www.thrombosisjournal.com/content/pdf/1477-9560-4-15.pdf [ Links ]
6. Walker P, Gregg A. Screening, Testing, or Personalized Medicine: Where do Inherited Thrombophilias Fit Best? Obstet Gynecol Clin N Am 2010; 37(1):87–107. Acceso 26 de julio 2014. Disponible en: http://www.sciencedirect.com/science/article/pii/S0889854510000227
7. Román González A, et. al. Paciente con deficiencia de proteína C y múltiples trombosis: reporte de caso. Iatreia 2007; 20(3):308-13. Acceso 17 de junio 2014. Disponible en: http://aprendeenlinea.udea.edu.co/revistas/index.php/iatreia/article/view/4411/3920 [ Links ]
8. Anderson J, Weitz J. Hypercoagulable States. Clin Chest Med 2010; 31(4);659–73. Acceso 15 de junio 2014. Disponible en: http://www.chestmed.theclinics.com/article/S0272-5231(10)00085-7/abstract
9. Swamy N, Vanitha N, Shashikala P. Protein C and Protein S Deficiency in Thrombogenic Events. Journal of Evolution of Medical and Dental Sciences 2013; 2(46):9054-58. Acceso 7 de agosto 2014. Disponible en: http://www.jemds.com/data_pdf/Shashikala.pdf
10. Lipe B, Ornstein DL. Deficiencies of Natural Anticoagulants, Protein C, Protein S, and Antithrombin. Circulation 2011; 124(14):365-68. Acceso 20 de junio 2014. Disponible en: http://circ.ahajournals.org/content/124/14/e365.full.pdf+html [ Links ]
11. Zamora-González Y, Agramonte-Llanes O, Rodríguez-Pérez L. Deficiencia de proteínas C y S: marcadores de riesgo trombótico. Rev Cubana Hematol Inmunol Hemoter 2013; 29(1):40-7. Acceso 14 de junio 2014. Disponible en: http://scielo.sld.cu/scielo.php?pid=S0864-02892013000100005&script=sci_arttext [ Links ]
12. Wypasek E, Anetta U. Protein C and Protein S Deficiency - Practical Diagnostic Issues. Adv Clin Exp Med 2013; 22(4):459–67. Acceso 13 de Julio 2014. Disponible en: http://www.advances.am.wroc.pl/pdf/2013/22/4/459.pdf
13. Kitchens C, et.al. Thrombotic Storm Revisited: Preliminary Diagnostic criteria Suggested by the Thrombotic Storm Study Group. Am J Med 2011; 124(4):290-96. Acceso 19 de agosto 2014. Disponible en: http://www.amjmed.com/article/S0002-9343(10)01043-0/pdf [ Links ]
14. Thornsberry LA, LoSicco KI, English JC. The skin and hypercoagulable states. J Am Acad Dermatol 2013; 69(3):450-62. Acceso 22 de agosto 2014. Disponible en: http://www.jaad.org/article/S0190-9622(13)00211-9/pdf [ Links ]
15. Pierangeli S, et.al. Acquired and Inherited Thrombophilia Disorders in Pregnancy. Obstet Gynecol Clin N Am 2011; 38(2):271–95. Acceso 3 de agosto 2014. Disponible en: http://www.sciencedirect.com/science/article/pii/S0889854511000295
16. Carbone JF, Rampexad R. Prenatal Screening for Thrombophilias: Indications and Controversies. Clin Lab Med 2010; 30(3):747–60. Acceso 1 de agosto 2014. Disponible en: http://www.sciencedirect.com/science/article/pii/S0272271210000533
17. Margeti S. Laboratory investigation of thrombophilia. J Med Biochem 2014; 33(1):28-46. Acceso 8 de agosto 2014. Disponible en: http://www.dmbj.org.rs/jmb/pdf/2014-1/5.pdf [ Links ]
18. Nicolaides A. The Case Against Routine Screening for Thrombophilia. Dis Mon 2010; 56(10): 569-73. Acceso 26 de Julio 2014. Disponible en: http://www.diseaseamonth.com/retrieve/pii/S0011502910001112 [ Links ]
19. Khor B, Van Cott E. Laboratory Evaluation of Hypercoagulability. Clin Lab Med 2009; 29(2): 339–66. Acceso 10 de julio 2014. Disponible en: http://www.sciencedirect.com/science/article/pii/S0272271209000134
20. Cohoon K, Heit J. Inherited and Secondary Thrombophilia. Circulation 2014; 129(2):254-7. Acceso 20 de agosto 2014. Disponible en: http://circ.ahajournals.org/content/129/2/254.full.pdf+html [ Links ]
21. Middeldorp S. Evidence-based approach to thrombophilia testing. J Thromb Thrombolysis 2011; 31(3):275–81. Acceso 16 de julio 2014. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3056012/pdf/11239_2011_Article_572.pdf
22. Baglin T, et. al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149(2):209–20. Acceso 25 de agosto 2014. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2009.08022.x/pdf
23. Galioto NJ, Danley DL, Van Maanen RJ. Recurrent Venous Thromboembolism. Am Fam Physician 2011; 83(3):293-300. Acceso 27 de julio 2014. Disponible en: http://www.aafp.org/afp/2011/0201/p293.pdf [ Links ]
24. Silvey M, Carpenter S. Inherited Thrombophilia in Children. Curr Probl Pediatr Adolesc Health Care 2013; 43(7):163-68. Acceso 13 de julio 2014. Disponible en: http://www.cppah.com/article/S1538-5442(13)00065-5/abstract [ Links ]
25. Maqbool S, et. al. Protein-C deficiency presenting as pulmonary embolism and myocardial infarction in the same patient. Thromb J 2013; 11:19. Acceso 21 de junio 2014. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3849523/pdf/1477-9560-11-19.pdf

