










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Científica Ciencia Médica
versión impresa ISSN 1817-7433versión On-line ISSN 2220-2234
Rev Cient Cienc Méd v.17 n.2 Cochabamba 2014
ARTÍCULO ORIGINAL
Impacto del Síndrome de West en pacientes del Hospital del Niño Manuel Ascencio Villarroel
Impact of West Syndrome in Patients from Manuel Ascencio Villarroel's Children Hospital
Cinthya Andia Berazain1, Dra. Heydi Sanz Arrazola2
1 Facultad de Medicina "Dr. Aurelio Melean", Universidad Mayor de San Simón, Cochabamba, Bolivia.
2 Neurología-Pediátrica. Docente de la Cátedra de Pediatría en la Facultad de Medicina UMSS.
Correspondencia a: Cinthya Andia Berazain andiaberazain_cinthya@hotmail.com
Procedencia y arbitraje: no comisionado, sometido a arbitraje externo.
Recibido para publicación: 31 de julio de 2014
Aceptado para publicación: 19 de noviembre 2014
Citar como: Rev Cient Cienc Med 2014;17(2):9-13
Abreviaturas utilizadas en este artículo:
SW = Síndrome de West
EEG = Electroencefalograma
ILAE = Liga internacional contra la epilepsia
CRH = Hormona liberadora de corticotropinas
TC = Tomografía computarizada
RM = Resonancia magnética
VGB = Vigabatrina
ITU: Infecciones del Tracto Urinario
RESUMEN
El síndrome de West es una encefalopatía epiléptica pediátrica dependiente de la edad caracterizada por la tríada clásica de: espasmos epilépticos, patrón hipsarrítmico y retraso mental. Se inicia en la mayoría de los pacientes durante el 1er año de vida, con una incidencia entre los 3 y 12 meses de edad. Presenta varias etiologías: criptogénica, idiopática y sintomática.
El objetivo de la investigación es describir la presentación clínica del Síndrome de West en nuestro medio, el diagnóstico y el tratamiento empleado.
El estudio realizado es de tipo observacional y de corte transversal. El universo comprende 377 pacientes menores de 2 años con Epilepsia que acudieron al consultorio de Neurología Pediátrica del Hospital del Niño Manuel Ascencio Villarroel del Complejo Hospitalario Viedma de Cochabamba-Bolivia, del 1ero de enero de 2010 a 31 de diciembre de 2013.
La Muestra son los 12 pacientes que fueron diagnosticados con Síndrome de West.
En los resultados se destaca: que los 12 pacientes cumplían con la triada clásica de la enfermedad, siendo el 100% sintomático, correspondiendo al género femenino 8 y 4 al masculino.
La edad en la que se hizo el diagnóstico con mayor frecuencia fue de 9 a 12 meses. Se realizó una evaluación de los antecedentes perinatales y neonatales entre los que se destacan: 5 casos con embarazo pre término y 6 casos con asfixia perinatal.
Se realizaron pruebas diagnósticas complementarias con: tomografía axial computarizada y electroencefalograma.
Se concluye que la presentación clínica del Síndrome de West en nuestro medio es sintomática en los 12 casos encontrados, el diagnostico se basó en la clínica y exámenes complementarios. El tratamiento principalmente empleado es el ácido valproico, debido a que la Hormona Adenocorticotrópica no está disponible en nuestro país.
Palabras clave: Síndrome de West, espasmos infantiles, hipsarritmia
ABSTRACT
West syndrome is a pediatric epileptic encephalopathy dependent on age characterized by the classic triad of epileptic spasms and mental retardation hypsarrhythmic pattern. It begins in most patients during the 1 st year of life, with an incidence between 3 and 12 months of age. Presents various etiologies: cryptogenic, idiopathic and symptomatic.
The aim of the research is to describe the clinical presentation of West syndrome the diagnosis and treatment used.
The study is descriptive and cross-sectional.The universe comprises 377 patients younger than 2 years with epilepsy who attended the clinic of Pediatric Neurology Children's Hospital Manuel AscencioVillarroelViedma Hospital in Cochabamba, Bolivia, January 1, 2010 to December 31,2013. The shows are the 12 patients who were diagnosed with West syndrome.
In the results emerged: the 12 patients met the classic triad of the disease, with 100% symptomatic, corresponding to 8 and 4 female to male.
The age at which the diagnosis is most often made was 9 to 12 months. 5 cases with preterm pregnancy and 6 cases with perinatal asphyxia:evaluation of prenatal and neonatal history including highlights was performed. Computed tomography and electroencephalogram: Additional diagnostic tests were performed. It is concluded that the clinical presentation of West syndrome in our country is symptomatic in 12 cases found, the diagnosis was based on clinical and complementary examinations. Treatment is mainly used valproic acid because the adrenocorticotropic hormone is not available in our country.
Keywords: West syndrome, spasms ¡nfantile, hypsarrhythmia
INTRODUCCIÓN
El Síndrome de West (SW) es una encefalopatía epiléptica de la infancia temprana asociada a diversos factores causales, se caracteriza por la triada clásica de: espasmos infantiles, retardo o detención del neurodesarrollo y el patrón hipsarrítmico del electroencefalograma (EEG)1,2,3,4,5,6,7,8,9,10,11.
En el año 1841 el médico ingles Willian James West (1793-1848) describió "una forma peculiar de convulsiones del lactante", que presentaba su hijo de 4 meses de edad, en la Revista Lancet. Posteriormente se evidenció que la mayoría de los pacientes con espasmos infantiles tienen cierto grado de retraso mental3,7,8,9,10.
El patrón hipsarrítmico EEG fue descrito por Gibbs y Gibbs en 1952. A partir de 1960 la triada clásica de esta patología, fue denominada S.W10.
Afecta 1 de cada 4-6.000 niños para 1960, informes recientes del año 2003 señalan una incidencia de 1/.2000 a 4.000 niños, representando el 47% de las epilepsias del primer año de vida1,3. Se estima su prevalencia de 0,15 a 0,2 por cada 1.000 en niños menores de 10 años3. Desde el punto de vista de la etiopatogenía y por la Liga Internacional Contra la Epilepsia (ILAE-1989) el S.W se divide en: criptogénico (10-15%) sin causa aparente y sintomático (60-90%), por múltiples causas como: trastornos prenatales (más frecuentes), perinatales o postnatales. Su frecuencia oscila entre el 2-10% de todos los casos de epilepsia infantil y representa la forma más frecuente de epilepsias del primer año de vida (excluyendo las convulsiones neonatales y las crisis febriles) 1,3,4,6,7,9,10,11
En 1991 la ILAE definió el S.W idiopático (5%).-pacientes con desarrollo psicomotor normal, antecedentes perinatales negativos, ausencia de convulsiones y estudios de neuroimagen normales3,10.
La edad de inicio del S.W es entre los 3 a 12 meses con alta incidencia entorno al 5to a 6to mes 1,4,10. Tiene un leve predominio en varones (1,5-1) representando un 60%1,3,6,7,10.
La fisiopatología del SW se desconoce, se han postulado varias hipótesis para intentar explicarla como: daño a un cerebro inmaduro, falta de equilibrio entre los neurotransmisores del tallo cerebral, zonas de perfusión anormal y anomalías del sistema inmunitario. Sin embargo la hipótesis actual más aceptada es la sobreproducción de la hormona liberadora de corticotropinas(CRH) que provoca hiperexcitabilidad neuronal y crisis convulsivas2,3,7,8,9,10,11.
Las manifestaciones clínicas comprenden espasmos epilépticos, que generalmente son: bilateral, simétrica y bifásica; acompañados de una breve pérdida de conciencia. Existen tres tipos principales de espasmos: en flexión, extensión (menos comunes) o mixtos (más comunes), simétricos o asimétricos3,4,7,10,11. Los factores que pueden precipitarla son: ruidos repentinos, la estimulación táctil, el hambre y el excesivo calor ambiental; gran número de pacientes con SW presentan retardo psicomotor antes del inicio de los espasmos 11.
El diagnóstico se sospecha por la clínica, pero a veces el cuadro clínico puede confundirse con: anomalías funcionales, y otros problemas neurológicos de base. El EEG se realiza en todos los casos, caracterizado por puntas y ondas lentas, ondas de perfil escarpado, de gran amplitud desordenadas que varían de un momento a otro tanto en duración como en localización, esencial para la confirmación de la enfermedad al observar el patrón de hipsarrítmia. Otras técnicas de neuroimagen, son: tomografía computarizada (TC), ecografía transfontanelar y resonancia magnética (RM)3,4,5,7,10,11.
Las guías actuales para el tratamiento del SW proponen un plan farmacológico divido en tres líneas de elección. La ACTH y el ácido valproico medicamentos de primera línea, la vigabatrina de segunda línea (primera línea en casos de esclerosis tuberosa y enfermedades metabólicas) otros son: la prednisona, acido valprico (su efectividad varia del 40 al 73%), los fármacos de tercera línea utilizados si persisten los espasmos, son: las benzodiacepinas como nitrazepam, clonazepam (que no son muy utilizadas por los efectos adversos), el topiramato, tropigina y la lamotrigina. En los casos en que no hay respuesta al tratamiento farmacológico o se encuentra contraindicado, se plantea la posibilidad de un abordaje quirúrgico para extirpar la zona de lesión cerebral1,3,7,8,9,10,11.
Las dosis utilizadas de ACTH varían de 20 a 40 unidades hasta 120 a 160 unidades por día. La duración del tratamiento varía de 2, 4 a 8 semanas. Las complicaciones de la corticoterapia, sobre todo de la ACTH son frecuentes (37% de los pacientes). La vigabatrina (VGB) se emplea a dosis de 80 a 200 mg/ kg/dia en dos dosis4,9.
Se recomienda también que el infante reciba estimulación temprana y terapia de rehabilitación de acuerdo con su edad10.
El pronóstico del S.W depende si el paciente inicialmente se clasifica como: idiopático, criptogénico o sintomático, el pronóstico es mejor en los casos criptogénicos e idiopáticos. En los casos criptogénicos la demora en el comienzo del tratamiento se asocia con un pronóstico peor. En los casos sintomáticos el pronóstico suele ser más severo2,3,4,9,10,11.
El retraso psicomotor ocurre en el 90% de los casos de S.W, asociado con frecuencia a trastornos de conducta y rasgos autistas. El 55 a 60% de los pacientes desarrollan otros tipos de epilepsia como el síndrome de Lennox-Gastaut y epilepsias con crisis parciales complejas. La mortalidad de este síndrome es del 5%3,7,10,11.
El pronóstico de los niños con S.W y Síndrome de Down parece ser mejor que el de los niños con S.W en la población general3,10.
La importancia del presente trabajo de investigación radica en mostrar una patología poco frecuente en nuestro medio como es el Síndrome de West que conlleva a errores en el diagnostico debido a que se subestima su presencia. El diagnóstico a temprana edad es imprescindible para que estos pacientes reciban el tratamiento precoz para así reducir las complicaciones, y tengan un pronóstico favorable y mejor calidad de vida.
Se planea que la investigación sirva a los profesionales y estudiantes del área de la salud, para que tengan en consideración la presentación de este Síndrome en nuestro medio, la clínica, el diagnóstico y tratamiento; así como la importancia de realizar una adecuada exploración e historia clínica, no dejando de lado los antecedentes prenatales, perinatales y neonatales en estos pacientes, que son datos relevantes.
MATERIALES Y MÉTODOS
El estudio que se realizó es de tipo descriptivo y de corte transversal.
El universo comprende los 377 pacientes menores de 2 años con Epilepsia que acudieron al consultorio de Neurología Pediátrica del Hospital del Niño Manuel Ascencio Villarroel del Complejo Hospitalario Viedma de Cochabamba-Bolivia, del 1ero de enero de 2010 a 31 de diciembre de 2013.
Muestra: comprenden 12 pacientes que fueron diagnosticados con S.W del 1ero de enero de 2010 a 31 de diciembre de 2013.
Criterios de inclusión: pacientes con diagnóstico de Epilepsia (crisis tipo espasmos masivos) y con diagnóstico de S.W menores de 2 años.
Criterios de exclusión: pacientes otros tipos de epilepsia.
Se revisaron retrospectivamente con historias clínicas de pacientes con diagnóstico de epilepsia y con diagnóstico de S.W. Se analizó la frecuencia y los factores asociados presentes en los pacientes. Se obtuvieron los datos de edad de diagnóstico del S.W, antecedentes perinatales y neonatales, exámenes de gabinete como neuroimagen y tratamiento. Posteriormente los datos encontrados fueron registrados y tabulados, para ello se empleó el programa de Microsoft Office Excel 2010® para el procesamiento y computo de los mismos.
Las variables a estudiar son: sexo, edad de inicio, antecedentes perinatales, antecedentes neonatales, tipos de espasmos, anomalías en la exploración neurológica, comorbilidad no neurológica, alteraciones en la neuroimagen, tratamiento médico utilizado.
RESULTADOS
Durante el periodo en estudio se encontró que los 12 pacientes cumplían con la triada clásica del S.W, y son de etiología sintomática; correspondiendo al género femenino 8 y 4 al género masculino.
La edad en la que se hizo el diagnóstico de S.W con mayor frecuencia fue de 9 a 12 meses. (Ver Tabla Nº 1)

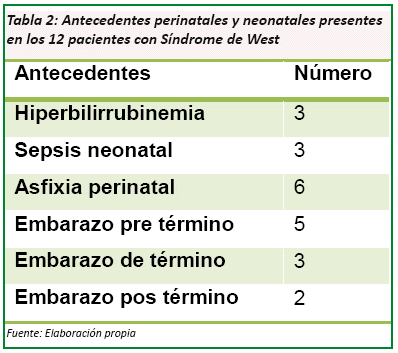
Dentro de los antecedentes perinatales y neonatales se destacan: 5 casos con embarazo pre término, 6 casos con asfixia perinatal y 2 casos con embarazo pos termino entre los destacados. (Ver Tabla Nº 2)
En cuanto a los tipos de espasmos en los 12 casos se caracterizó por presentar: pacientes que cursaban con mioclonias breves en flexión, espasmos frecuentes en salvas con alta frecuencia y retardo en el desarrollo psicomotor.
Las anomalías a la exploración neurológica fueron:
De los 12 casos evaluados 11 presentaron retardo en el desarrollo psicomotor al momento del diagnóstico y 1 paciente curso con desarrollo psicomotor dentro de los parámetros normales para su edad.
De los pacientes con retraso en la exploración neurológica se encontró: 10 pacientes con hipotonía axial, hipertonía en las 4 extremidades y con espasticidad en tijera hiperrefléxica, y tetraparesia espastica y 1 paciente con hemiparesia derecha. El paciente que presentaba desarrollo psicomotor normal no presentaba ninguna alteración al examen neurológico.
Las comorbilidades no neurológicas encontradas en algunos pacientes: desnutrición, anemia, TORCH, síndrome nefrótico, criptorquidia, ITU, hidrocele e hipoglicemia.
Las alteraciones en la Neuro-imagen fueron:
La tomografía axial computarizada(TAC) se realizó solo en dos casos donde se reportó: en el primer caso atrofia cerebral difusa con infarto cerebral frontal periventricular y en el segundo caso atrofia cerebral. No se pudo completar el estudio de imagen en todos los pacientes porque no contaban con recursos.
En el electroencefalograma (EEG) se encontró 8 pacientes con hipsarrítmia, uno con polipunta onda lenta, dos con actividad interhemisferica, y uno con asimetría interhemisferica.
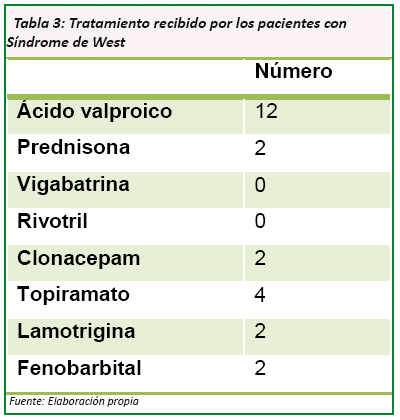
El tratamiento principalmente empleado en los 12 casos fue con ácido valproico. Este tratamiento se combinó con prednisona en 2 casos, con topiramato en 4, clonacepam en 2 casos y lamotrigina en 2 casos. (Ver Tabla Nº 3) y fenobarbital en 2 casos.
DISCUSIÓN
La edad de presentación del S.W se encuentra entre los tres y los 12 meses, con alta incidencia entre los cinco a seis meses1,4,10. En la investigación se encontró que la edad de diagnóstico más frecuente fue de 9 a 12 meses (66,6%). Esto probablemente a que muchas veces el cuadro clínico puede confundirse con anomalías funcionales, y otros problemas neurológicos. El diagnóstico de los 12 pacientes con S.W fue después del inicio más o menos 1 mes de los espasmos, lo que plantea un diagnóstico tardío de esta patología.
El S.W tiene un leve predominio en varones (1,5-1) representando un 60%1,3,6,7,10. Sin embargo en el estudio realizado se encontró: que de los 12 pacientes, una mayor cantidad de casos corresponden al género femenino 8 (66,6%) y 4 al género masculino (33,3%).
La etiopatogenia del S.W la divide en: sintomática y criptogénica. En el S.W sintomático (60-90%) las crisis son resultado de lesiones estructurales cerebrales identificables. Las causas de este padecimiento pueden ser trastornos prenatales (más frecuentes), perinatales o postnatales 3,10,11.
Dentro de los antecedentes perinatales y neonatales de los 12 pacientes se encontró: 5 casos (41,6%) por embarazo pre término, y 6 casos (50%) con asfixia perinatal. Una complicación que se tuvo en el estudio fue que los antecedentes prenatales pese a que son los más frecuentes, como causas en esta patología, no se pudieron obtener ya que las historias proporcionadas estaban incompletas, por lo que se las descarto para el estudio tomando en cuenta solo los antecedentes perinatales y neonatales.
El EEG se caracteriza por el patrón hipsarrítmico característico de esta patología 3,4,5,7,10,11. En el EEG que se realizó a los 12 pacientes se encontró 8 pacientes con hipsarrítmia (66,6%), uno con polipunta onda lenta (8,3%), dos con actividad interhemisferica (16,6 %), y uno con asimetría interhemisférica (8,3 %). Los estudios de neuroimagen que se emplearon en los pacientes fueron: el TAC que se realizó solo en dos casos (16,6%) donde se reportó: atrofia cerebral difusa con infarto cerebral frontal periventricular, en un caso y en otro caso atrofia cerebral. Esto debido a que gran parte de los familiares de los pacientes con diagnóstico de S.W son de ingresos económicos reducidos como para pagar este tipo de estudios, ya que el seguro no lo cubre.
El tratamiento principalmente empleado es el ácido valproico. Este tratamiento se combinó con prednisona, con topiramato, clonacepam, fenobarbital o lamotrigina, dependiendo de las características clínicas de cada paciente y su tolerancia; debido a que la hormona adenocorticotropica (ACTH) pese a que es el medicamento de primera línea en la terapéutica farmacológica del S.W, no está disponible en nuestro país.
Se concluye que la presentación clínica del Síndrome de West en nuestro medio es de etiología sintomática en los 12 casos encontrados, el diagnostico se basó en la clínica, el electreoencefalograma (EEG), tomografía axial computarizada (TAC) y ecografía transfontanelar. Esta última no se efectuó en todos los pacientes, por lo que no se mencionó en el estudio. El tratamiento principalmente empleado en nuestro medio es el ácido valproico, que se administra en combinación con otros medicamentos (dependiente de la presentación clínica del paciente, su tolerancia), debido a que la Hormona Adenocorticotropica no está disponible en nuestro país.
REFERENCIAS
1. Arce-Portillo E, Rufo-Campos M, Muñoz-Cabello B, Blanco-Martínez B, Madruga-Garrido M, Ruiz-Del Portal L, et al. Síndrome de West: etiología, opciones terapéuticas, evolución clínica y factores pronósticos. Rev Neurol(En linea) 2011; 52: 81-9. Disponible en: http://www.neurologia.com/pdf/Web/5202/bf020081.pdf Fecha de acceso: 03-05-2014
2. Travieso-Tellez A, Lantigua-Cruz A, García-García R. Estudio clínico genético de pacientes cubanos con Síndrome de West. Rev Ciencias Médicas (En línea) Mar- Abr 2012; Vol. 16 no. 2 Disponible en: http://scielo.sld.cu/scielo.php?pid=s156131942012000200005&script=sciarttext Fecha de acceso: 03-05-2014
3. Atuesta A, Reina D. C, Lozano W, y Gélvez X. Síndrome de West: encefalopatía epiléptica. Rev Medicas UIS (En línea) 2009; vol. 22, no 1. Disponible en: http://132.248.9.34/hevila/MedicasUIS/2009/vol22/no1/7.pdf Fecha de acceso: 03-05-2014
4. Bauzano-Poley E, Rodriguez-Vives M. A, y Rodriguez-Barrionuevo A. C. Epilepsias y síndromes epilépticos del lactante. Hospital materno infantil, Málaga Asociación Española de Pediatría. Protocolos actualizados 2008 Disponible en: www.aeped.es/protocolos/http://www.aeped.es/sites/default/files/documentos/3-epilepsialactante.pdf Fecha de acceso: 03-05-2014
5. Fejerman N, Diagnósticos diferenciales del Síndrome de West. Rev Neurol (En línea) 2013; 57 (Supl 1): S125-8 Disponible en: http://www.neurologia.com/pdf/Web/57S01/bkS01S125.pdf Fecha de acceso: 07-05-014
6. Toro-Alonso V. El juego en alumnos con necesidades educativas especiales: Síndrome de West y otras Encefalopatías Epilépticas. Rev de educación inclusiva (En línea) 2012; vol. 6, p. 72-87. Fecha de acceso: 11-05-2014
7. Pérez A. H., Sanchéz M. B., Fernández R. C., Serrano B. R. Síndrome de West. Estudio Clínico, Neurofisiológico y de Neuroimagen en las Tunas. Innovación Tecnológica (En línea) 2009; vol. 15, no 1. Disponible en: http://innovaciontec.idict.cu/innovacion/article/view/178/179 Fecha de acceso: 17-05-2014
8. Universidad de Francisco Marroquín Facultad de Medicina. Síndrome de West 2008. disponible en: http://medicina.ufm.edu/index.php/S%C3%ADndrome_de_West Fecha de acceso: 18-05-2014
9. Asociación Andaluza de Epilepsia. Descripción del Síndrome de West. 30 Jun. 2014. Disponible en: http://www.apiceepilepsia.org/Sindrome-de-West-o-Espasmos-Infantiles
Fecha de acceso: 25-05-0214
10. Morón-García G. del C, Urrutia-Torres F. J, y Fuentes-Cuevas M. del C. Frecuencia y antecedentes asociados con el síndrome de West. Arch Inv Mat Inf. (En linea) Ene-abr 2012; Vol. IV, No. 1 pp. 7-12; Disponible en: http://www.medigraphic.com/pdfs/imi/imi-2012/imi121b.pdf Fecha de acceso: 01-06-2014
11. Matinez-Quezada D I. Síndrome West. Rev Médica MD(En linea) jul-sept 2010;vol. 2, p. 1. Disponible en: http://www.revistamedicamd.com/sites/default/files/revistas/rev_med_md_volumen_
2_numero_1_0.pdf#page=11 Fecha de acceso: 01-06-2014

