










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO Compartir

 Permalink
PermalinkRevista Científica Ciencia Médica
versión impresa ISSN 1817-7433versión On-line ISSN 2220-2234
Rev Cient Cienc Méd v.13 n.1 Cochabamba 2010
CASO CLÍNICO
Síndrome de Peutz-Jeghers y Obstrucción Intestinal Baja, Reporte de un Caso
Peutz-Jeghers Syndrome and Low Intestinal Obstruction, Report of a Case
Marielena Montero1, Alejandra V. Montoya Burgos1, Aleyda Muñoz1, Marlene Anaya Domínguez2
1 Estudiantes de Medicina, Universidad Mayor de San Simón. Cochabamba, Bolivia
2 Médico Cirujano Pediatra del Hospital Manuel Ascencio Villarroel Cochabamba, Bolivia
Correspondencia a: Alejandra V. Montoya Burgos alierobi@hotmail.com
Recibido para publicación: 9 de Julio de 2010
Aceptado para publicación: 14 de Agosto de 2010
Citar como: Rev Cient Cienc Med 2010; 13(1): 35-37
Abreviaciones y acrónimos utilizados en este artículo:
PAF = Poliposis Adenomatosa Familiar
PCF = Poliposis Colónica Familiar
APC = Adenomatous Poliposis Coli
RESUMEN
El presentamos el caso clínico de un niño de 12 años, que acude al Servicio de Pediatría del Hospital Manuel Ascencio Villarroel, transferido del Centro Pediátrico Albina Rodríguez de Patiño con los posibles diagnósticos de estreñimiento pertinaz, desnutrición de III grado secundario, Síndrome de Peutz-Jeghers probable, anemia microcítica e hipocrómica severa y soplo sistólico en estudio. Si bien el Síndrome de Peutz-Jeghers se presenta en contadas ocasiones en nuestro medio, el diagnóstico diferencial y sus complicaciones deben ser mejor estudiadas para así poder ser tratada de forma más oportuna. Consideramos importante este reporte porque en nuestro medio es una rara causa de abdomen agudo.
Palabras claves: Poliposis hamartomatosa intestinal, Lentiginosis perioral, Síndrome de pólipos y manchas.
ABSTRACT
The present case report of a child 12 year old boy, who was admitted to the pediatric department of the Hospital Manuel AscencioVillarroel, transferred to the Pediatric Center Albina Rodriguez Patiño with possible diagnoses of persistent constipation, grade III secondary malnutrition, Peutz-Jeghers probable syndrome, severe hypochromic microcytic anemia and systolic murmur in the study.While the Peutz-Jeghers syndrome rarely occurs in our environment, the differential diagnosis and its complications should be better studied so we can be treated in a more timely manner. We consider it important our report because this disease is a rare cause of acute abdomen in our environment.
Keywords: Polyposis, Hamartomatous Intestinal, Lentiginosis perioral, Polyps and Spots Syndrome
INTRODUCCIÓN
El síndrome de Peutz-Jeghers es un desorden autosómico dominante, caracterizado por la presencia de pólipos hamartomatosos intestinales y lesiones pigmentadas mucocutáneas que también pueden encontrarse en palmas y plantas, la asociación entre Síndrome de Peutz-Jeghers y cáncer ha sido una controversia a lo largo de las décadas pasadas. Desde los años ochenta ala actualidad, interesantes estudios revelaron incremento del riesgo de desarrollo de cáncer en pacientes afectados y no afectados de familias con síndrome de Peutz-Jeghers, localizándose en sitios gastrointestinales y extraintestinales1,2,3.
La incidencia estimada del síndrome de Peutz-Jeghers es entre 1 en 8300 a 1 en 29000 nacidos vivos, la edad de diagnóstico varía entre los 9 y 39 años de edad siendo el promedio de 29 años1,2.
Se debe realizar diagnóstico diferencial con otros síndromes de poliposis de tipo adenomatosa, incluyendo la poliposis adenomatosa familiar, el síndrome de Cowden y el Síndrome de Bannayan-Riley-Ruvalcaba2.
PRESENTACIÓN DEL CASO
Paciente de sexo masculino, de 12 años de edad, procedente de Chapare, Cochabamba; es internado en el Hospital Manuel Ascencio Villarroel, con un cuadro clínico de aproximadamente 14 días de evolución, caracterizado por presentar: dolor abdominal de tipo cólico intermitente en mesogastrio, de elevada intensidad acompañado de vómitos en varias oportunidades, falta de eliminación de gases y heces con antecedente de cuadros similares desde hace 2 años de aparente resolución espontánea. Dicho cuadro se exacerbó hace mas o menos 10 días atrás por lo que los padres lo llevaron al Hospital Albina Rodríguez de Patiño donde se lo hospitalizó con el diagnóstico de impactación fecal, a su ingreso presentaba palidez de piel y mucosas, con datos clínicos, antropométricos y laboratoriales de desnutrición grado II, presencia de soplo tricuspídeo de II/VI de intensidad, abdomen blando depresible doloroso a la palpación superficial y profunda. Dentro del manejo se le realizó una proctoclisis, ante la mala respuesta y no observar mejoría el paciente es transferido al hospital Manuel Ascencio Villarroel, con diagnóstico de Dolicocolon y posible Síndrome de Peutz-Jeghers.
Examen Físico: Paciente en mal estado general, en decúbito dorsal pasivo, consciente y orientado en las tres esferas, afebril, fascie álgida, piel y mucosas pálidas, ligeramente secas, peso de 25 kilogramos, con signos vitales: Frecuencia respiratoria de 26 por minuto; Temperatura de 37°C; Pulso de 94 por minuto, regular amplio; Presión arterial de 100/64 mmHg.
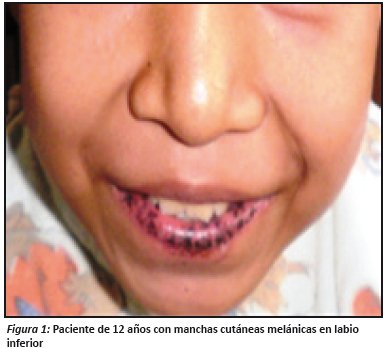
A la exploración física se encontraron pequeñas zonas puntiformes hiperpigmentadas en labio inferior (ver fig.1).
Tórax simétrico, enflaquecido, con expansibilidad y elasticidad conservadas, ruidos cardíacos de ritmo regular, normofonéticos, soplo sistólico funcional en foco tricuspídeo de grado II-VI.
Abdomen blando, plano, doloroso a palpación, con RHA (+), hiperactivos (de lucha). Se palpa una masa móvil, de más o menos 11cmx 7 cm. de diámetro a nivel de flanco izquierdo y al tacto rectal se encuentra ampolla rectal vacía.
La impresión diagnóstica de inicio fue de abdomen agudo por obstrucción intestinal baja, anemia clínica grave, desnutrición II grado y probable síndrome de Peutz-Jeghers.
Interconsultas y Estudios Pendientes:
Hemograma: eritrocitos; 3.200.00/mm3,, Hematocrito 19,8%, Leucocitos 6.700/mm3
Electrocardiograma. Ritmo sinusal, con eje de +60.
Interconsulta con cardiologia. Hemodinámicamente compensado, se sugiere vigilancia cardiopulmonar durante el acto.
Interconsulta con anestesiología. Recomienda reservar dos paquetes de concentrado globular.
Tratamiento y evolución
El paciente fue intervenido quirúrgicamente con el diagnóstico preoperatorio de masa tumoral en estudio. Se realizó una laparotomía exploradora y los hallazgos fueron dos masas tumorales en lumen de intestino delgado yeyuno ileal e íleon colónico. Se realizó resección de los segmentos con anastomosis término-terminal.
Egresó con el diagnóstico de invaginación ileocólica e ileoyeyunal (doble invaginación). Se realiza estudio de muestras de segmentos reseccionados, líquido peritoneal y ganglio para examen patológico que confirma la presencia de pólipos hamartomatosos.
El paciente en buen estado general afebril, hidratado, mucosas húmedas y rosadas, fue dado de alta en su día 11 de internación y día 9 post quirúrgico con el diagnóstico de Síndrome de Peutz-Jeghers, con doble invaginación.
DISCUSIÓN
El Síndrome de Peutz-Jeghers se diagnostica con mayor frecuencia entre los 9 y 39 años2.
Se transmite como un patrón autosómico dominante de herencia.
La presentación clásica comprende: pólipos gastrointestinales que son hamartomas típicos, su histología se caracteriza por extensiones cubiertas de músculo liso que surge de la muscularis mucosae y se extiende al interior del pólipo, dando apariencia circunvolucionada. Esta capa está cubierta por mucosa intestinal normal que contiene todos los elementos, incluyendo epitelio columnar absortivo, células de Paneth, argentafines y caliciformes. En orden decreciente de frecuencia se presenta en yeyuno, íleon, colon, recto, estómago, duodeno y apéndice. Los pólipos del intestino delgado y colon tienden a ser pedunculados, mientras que los del estómago son sésiles2,8.
Ya que los lugares de presentación son variables en cada paciente; el reporte de cirugía precisa que se encontraron pólipos hamartomatosos en Íleon-yeyunal e Íleon-colónico, no diferenciados en la radiografía y ecografías por la acumulación de heces.
Hiperpigmentación mucocutánea, representada por pequeñas máculas de color café de 1 a 5 mm de diámetro que se observan en los labios en un 96% de los casos, seguida de la pigmentación de la mucosa oral en el 83%, pueden presentarse en las encías y el paladar, pero no en la lengua, áreas alrededor de la nariz y los ojos están involucrados en un 36%, las extremidades en un 32%, en especial en la palma y dedos de la mano. Ocasionalmente también pueden presentarse en los genitales, planta, dedos de los pies y la mucosa intestinal1,2,9.
La anemia se produce por hemorragia intestinal baja, producto del sangrado de los pólipos10; además de los valores bajos de hemoglobina y hematocrito, se presentó una microcitosis, hipocromía y anisocitosis leve, lo que demuestra la presencia de anemia, siendo necesarias transfusiones de concentrado de glóbulos rojos, para restablecer el equilibrio hemodinámico del paciente.
Es típica la obstrucción intestinal parcial caracterizada por ataques recurrentes de dolor abdominal cólico consecutivo a intususcepción transitoria, es poco común que haya obstrucción completa y casi todos los episodios de intususcepción se resuelven de forma espontánea1,13; en este paciente la intususcepción produjo una obstrucción completa, presentandose además como doble invaginación.
Patologías con las que se debe hacer diagnóstico diferencial son la PAF o PCF que es una enfermedad hereditaria infrecuente con una incidencia de 1 caso / 10 000-20 000 habitantes. Se caracteriza por la aparición de numerosos pólipos adenomatosos gastrointestinales y por el desarrollo de cáncer colorrectal en prácticamente el 100% de los pacientes que no reciben un tratamiento adecuado. De forma característica aparecen más de 100 pólipos adenomatosos en el colon y recto.
La PAF clásica presenta un patrón de herencia autosómica dominante, el responsable es el gen APC situado en el cromosoma 5 (5q21)2. La mutación genética de este gen conduce a una mucosa hiper-proliferativa en todo el tracto intestinal. La poliposis adenomatosa familiar atenuada presenta en un 30% de los casos un patrón de herencia autosómica recesiva. No suelen tener hipertrofia retiniana ni tumores desmoides12.
Acerca de los procedimientos a emplear, son útiles las endoscopias altas y bajas para evaluación practicadas cada 2 años, se debe incluir valoraciones radiológicas de intestino delgado, laparotomías, siendo las polipectomias el manejo de elección para remoción de pólipos mayores de 1,5 cm, por otro lado, los pacientes deben ser sometidos a exámenes regulares de mama y ginecológicos11,12; la cirugía practicada fue una laparotomía exploradora con resección de los segmentos de intestino delgado, yeyuno ileal e íleon colónico y anastomosis término-terminal.
El pronóstico para las personas con síndrome de Peutz-Jeghers es bueno y la mayoría llega a edad adulta avanzada, aunque es raro, pueden morir por complicaciones como intususcepción, pérdida de sangre, cáncer y las operaciones necesarias del tratamiento1. En una revisión de Spiegelman el riesgo relativo de cáncer gastrointestinal en pacientes con el síndrome es de 13 veces el riesgo de la población común.
Esófago, estómago, intestino delgado, colon, recto, páncreas y vesícula biliar son los tumores gastrointestinales más comunes. De los tumores extragastro-intestinales asociados al síndrome de Peutz-Jeghers, cuyo riesgo de presentación es 15 veces mayor; el de mama es el mas común. Otros son el de pulmón, cuello uterino (adenocarcinoma), ovario y testículos (tumor de células de Sertoli), que no son muy frecuentes en la población14.
Sin bien esta patología no es muy bien estudiada en nuestro medio por la poca cantidad de casos que se presentan, su manejo en el Hospital Manuel Ascencio Villarroel fue acertado.
REFERENCIAS
1. Ashcraft Keith W, Murphy, Sharp, Sigalet, Snyder, Eric W, Fonkaisrud MD: Enfermedad intestinal inflamatoria y neoplasias del tubo digestivo. Murphy, Sharp, Sigalet, Snyder. Cirugía Pediátrica de Ashcraft. 3ra Ed. México, McGraw-Hill; 2002:193-195 [ Links ]
2. Abdo Francis J. M, Pérez Torres E, Bernal Sahagún F, Dzib Salazar J. Síndrome de Peutz-Jeghers. Rev. M.H.G.M. 2005;18(2): 99-105 [ Links ]
3. Pinto Sánchez J.P, Rebaza Vásquez S, Muñoz Mendoza S, Cárdenas V.M. Síndrome de Peutz-Jeghers y Adenocarcinoma. Rev.G.P. 2004; 40: 363-367 . [ Links ]
4. Ferraina P, Oria A, Hernández N: Fecaloma. Ferraina P, OriaA. Cirugía de Michans. 5ta ed. Buenos Aires, editorial el Ateneo; 2008: 865-867 . [ Links ]
5. Alonso Sánchez A, Benavides Orgaz M, Blanco Guillermo L, Brunet Vidal J, García Foncillas J, Mayordomo J.I, Pérez Segura P, Urioste Ascorra M, Blanco Guillermo I, González Romero S: Síndromes polipósicos-aspectos clínicos. Alonso Sánchez A, Benavides Orgaz M, Blanco Guillermo I, Brunet Vidal J, García Foncillas J, Mayordomo J.I, Pérez Segura P, Urioste Ascorra M. S.E.O.M-Cáncer hereditario. Madrid, Dispublic; 2006: 437-438.
6. Giardiello F.M, Brensinger J.D, Tersmette A.C, Goodman S.N, Petersen G.M, Booker S.V. Syndrome Peutz-Jeghers. S.H.C.C-Part II. 2005:307-316 [ Links ]
7. Ferreiro M, Harris P, Larraín F, Duarte I, Repetto G. Familia con síndrome de Peutz-Jeghers. Rev. C.P. 2000: 71(3): 370-4106 [ Links ]
8. Kumar V, Abbas A.K, Fausto N, Liu C. MD, PhD: Tracto gastrointestinal. Kumar V, Abbas A.K, Fausto N. Robbins y Cotran Patología Estructural y Funcional. 7ma ed. España, Elsevier; 2005: 862-863 [ Links ]
9. Rubio Tapia A., Ramírez Arias F, Ángeles Ángeles A, Uscanga L. Síndrome de Peutz-Jeghers. Rev G.M. 2005; 70(3):291-295 [ Links ]
10. Leung A.K.C, Wong A.L. Sangrado gastrointestinal bajo en niños. Rev. P.E.C. 2002;18(4): 319-323 [ Links ]
11. Weitz J.C, Berger Z, Sabah S, Silva H, Maiza Rodríguez E: Pólipos Colónicos. Weitz J.C, Berger Z, Sabah S, Silva H. Diagnóstico y Tratamiento de las Enfermedades Digestivas-Sociedad Chilena de gastroenterología. Santiago de Chile, IKU; 2002: 155-161. . [ Links ]
12. Plan oncológico comunidad valenciana, Lledó Matoses S, Payá Roma A: Otros síndromes de cáncer hereditario. Plan oncológico comunidad valenciana. Guía de práctica clínica en cáncer hereditario. 2da ed. Valencia, Generalitat Conselleria de Sanitat; 2009:99-103 [ Links ]
13. Pary Montecinos R. (Sociedad Boliviana de Cirugía), Salazar Peredo D. (Hospital La Paz): Intususcepción. Pary Montecinos R, Salazar Peredo D. INASES-Diagnóstico, tratamiento y procedimientos de emergencias en cirugía. Bolivia, Departamento técnico de salud INASES; 2008: 43-45 [ Links ]
14. John Ospina N, Álvaro Pío Q. Síndrome de Peutz-Jeghers. Presentación de casos y revisión de la literatura. Rev.C.G. 2009; 24(2): 1-21 [ Links ]

