











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El paraganglioma es un tumor neuroendocrino dependiente del sistema nervioso parasimpático que se encuentra generalmente en localización adrenal y extra-adrenal. Las localizaciones extra-adrenales son raras y ocurren más frecuentemente en el glomus carotideo.
En general son de mal pronóstico y tienen pobre respuesta a quimioterapia.
Presentamos el caso de una paciente con paraganglioma metastásico pulmonar que presento buena respuesta a terapia blanco con pazopanib.
PRESENTACIÓN DEL CASO
Mujer de 23 años de edad, con antecedente de tabaquismo y alcoholismo. Sin antecedentes oncológicos familiares. Presenta un cuadro clínico de tres meses de evolución con tos seca, disnea progresiva y cianosis por lo que acude un hospital de segundo nivel donde se realiza TAC de tórax con reporte de múltiples metástasis pulmonares.
En la consulta inicial, la paciente en silla de ruedas con aporte de oxígeno a 2l/minuto, taquicárdica, saturación de 84%. Con presencia de estertores crepitantes difusos bilaterales. Los paraclínicos de inicio Hb: 17, 6, GB 6300, Plaquetas: 301000, DHL: 312, AFP: 1.6, GCH:0.
Se realiza biopsia de nódulo sub-pleural izquierdo con reporte de neoplasia de células gigantes y poligonales compatible con paraganglioma. La inmunohistoquímica con CD 10, CK 20 y CKAE/AE3 negativos; S-100, cromogranina y sinaptofisina positivos.
Se realizan estudios de extensión con tomografía de tórax, abdomen y pelvis, en la que se evidencia enfermedad metastásica múltiple pulmonar.
Inicia tratamiento sistémico con esquema VAC con vincristina 2mg/m2 (dosis máxima 2mg),doxorrubicina: 75mg/m2, ciclofosfamida 1200mg/m2, recibió un total de 6 ciclos con enfermedad estable por tomografía y discreto beneficio clínico, pero dependiente de oxígeno.
Posteriormente presenta deterioro clínico con aumento de la disnea, tos, pérdida de peso, hiporexia y aumento de los requerimientos de oxígeno. Por tomografía de control, se evidencia progresión franca de la enfermedad a nivel pulmonar (Fig 1).
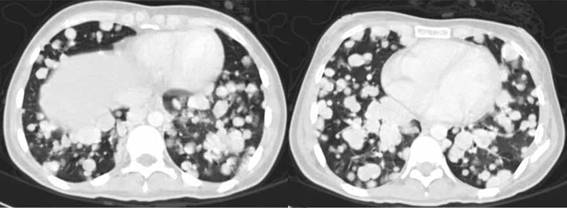
Se inicia terapia blanca con pazopanib a 800 mg por día por vía oral, con mejoría importante de la disnea al segundo ciclo de tratamiento y tras 6 ciclos con respuesta parcial por imagen. Actualmente lleva 12 meses de tratamiento con importante beneficio clínico, aumento de peso de 5 kilos, mejoría del estado funcional y sin dependencia del oxígeno. La ultima tomografía con respuesta parcial, pero disminución notable tanto del número como del tamaño de las lesiones pulmonares (Fig.2).
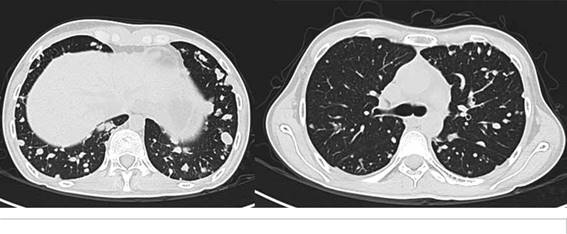
Tuvo buena tolerancia a pazopanib y el único evento adverso presentado fue hipotiroidismo grado 1 e hipopigmentación generalizada.
Actualmente en tratamiento sustitutivo con levotiroxina.
REVISIÓN DE LA LITERATURA
El término paraganglioma fue primeramente mencionado en 1908 por los patólogos Henri Alezais y Felix Peyron, los cuales notaron tumores con una reacción positiva a la cromafina en tejido adrenal y extra-adrenal. 2
Los paragangliomas son tumores neuroendocrinos que surgen a partir de células derivadas de paraganglios de la cresta neural y se distribuyen a lo largo del eje paravertebral y para-aórtico de la base del cráneo al suelo pélvico. 2
Afecta 2 a 5 personas por millón por año y tiene una prevalencia de 1:2500.
El pico de incidencia ocurre entre la tercera y quinta década de la vida. 2
Se ha descrito la regla de los “10” para paraganglioma/ feocromocitoma; con 10% de ocurrencia en el tejido extra-adrenal, 10% bilateral y 10% maligno. Sin embargo, de acuerdo a revisiones actuales, esta regla ya no es aplicada.
Aproximadamente 8% de los tumores esporádicos y 20 a 75% de los paragangliomas/ feocromocitomas hereditarias son bilaterales a la presentación; 5% de los tumores originados en adrenales y 33% de los extra-adrenales son malignos. 2
Alrededor del 40% de los pacientes con paraganglioma tiene mutaciones germinales en un conocido gen susceptible. (2) Un 25% de los paragangliomas pueden presentarse como parte de un síndrome hereditario: MEN2A, MEN2B, síndrome de Von Hippel Lindau y Neurofibromatosis tipo 1. 7
Esta enfermedad se presenta principalmente en las partes del cuerpo ricos en paraganglios, tales como la cabeza, el cuello, mediastino, glándula suprarrenal, y peritoneo posterior, incluso de la vejiga, el duodeno y tiroides. 8
Los paragangliomas extra-adrenales son raros y afectan principalmente al glomus carotideo. 1
En cuanto a la sintomatología, debido a la secreción de catecolaminas, los pacientes suelen presentar infarto de miocardio, arritmia, o un derrame cerebral, sin embargo, esto suele ser lo menos frecuente, siendo a menudo diagnosticados con una masa incidental totalmente asintomática. 2
Para el diagnóstico, las 2 modalidades más utilizadas de imagen en la valuación inicial son la tomografía computarizada y la resonancia magnética. Los estudios de medicina nuclear como MIBG (123 I-metayodobencilguanidina), 111-A-pentetreótido (exploración octreotida) y PET/ CT usando fluorodeoxiglucosa (FDG) y otros agentes radiomarcados son también utilizados para la localización de paragangliomas. 2
El tratamiento primario es la cirugía. En enfermedad metastásica, si es posible la resección, se debe considerar la citorreducción quirúrgica tanto del primario como de la metástasis. 2
En un estudio reciente de 287 pacientes con 221 pacientes con paraganglioma maligno, la supervivencia global a 5 años y la supervivencia específica de la enfermedad fueron de 80,0% y 86,4% respectivamente. 3
Muchos autores propusieron que paraganglioma es sensible a radioterapia, con altas tasas de control local y pocas complicaciones. 1
Sin embargo, la radioterapia puede ser considerada cuando existe progresión local de la enfermedad.
La enfermedad metastásica, tiene menos opciones de curación, a menos que pueda ser llevada a resección quirúrgica completa. Otros tratamientos investigados son la quimioterapia, 131I-MIBG, y / o la radiación que pueden ofrecer control de la enfermedad.
Dada la naturaleza a menudo indolente de la enfermedad, los tratamientos por lo general están reservados para los pacientes con progresión clara o síntomas graves. No hay estudios para dirigir el tiempo o el orden de los tratamientos. 2
La ciclofosfamida, vincristina y dacarbazina (CVD) es el régimen estándar de quimioterapia para el tratamiento de enfermedad metastásica, con tasas de respuesta alrededor de 50 a 60%, con respuesta completa del 4%, respuesta parcial del 37% y 14% de enfermedad estable. 4 Las respuestas tumorales usualmente se producen después de 2 a 4 ciclos de la terapia y la mediana de duración de la respuesta fue de 20 meses. Las toxicidades más comunes son la mielosupresión, neuropatía periférica y la toxicidad gastrointestinal. 4
Se ha descrito también tratamiento con temozolamida y metaiodobencilguanidina (MIGB); esta última debido al 6% de paragangliomas con avidez a MIGB. 2
En cuanto a terapia blanco, se han realizado varios estudios pequeños con inhibidores de mTOR (everolimus) el cual mostró pobres resultados. 2,5
Sunitinib, ha sido evaluado en un estudio retrospectivo de 17 pacientes con metástasis. Con una tasa de respuesta de 47% y mediana de SLP de 4,1 meses. 2,6
El pazopanib en un inhibidor multicinasa que ha mostrado respuestas favorables en el tratamiento de paraganglioma en reportes de casos.
Actualmente se está llevando a cabo un estudio de fase 2 del NCI con pazopanib en paraganglioma metastásico o recurrente que proporcionará una mejor información sobre la eficacia y seguridad de esta terapia dirigida.
CONCLUSIÓN
El paraganglioma es un tumor neuroendocrino dependiente del sistema nervioso parasimpático cuyo tratamiento primario es la cirugía. En el contexto metastásico, tiene pobre respuesta a quimioterapia y en general mal pronóstico. Sin embargo, emergen nuevas modalidades de tratamiento, entre ellas pazopanib que ha mostrado respuestas favorables en reporte de casos, sin embargo, aún se encuentra en fase de investigación, pero con resultados prometedores.

