










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica La Paz
versión On-line ISSN 1726-8958
Rev. Méd. La Paz vol.25 no.2 La Paz 2019
CASO CLÍNICO
MUCOPOLISACARIDOSIS TIPO VI: REPORTE DE TRES CASOS Y REVISIÓN DE LA LITERATURA
MUCHOPOLYSACCHARIDOSIS VI: REPORT OF THREE CASES AND LITERATURE REVIEW
Germán Meleán Gumiel1y2*, Valeria Aillón López 1, Gonzalo Taboada López 1
1. Instituto de Genética. Facultad de Medicina. UMSA. La Paz - Bolivia
2. Centro de Genética Molecular. La Paz - Bolivia
* Correspondencia al autor: g.melean@gmail.com
RESUMEN
El síndrome de Maroteaux-Lamy es una de las mucopolisacaridosis menos frecuente, con una incidencia aproximada de 0.36 a 1.30 por cada 100,000 nacidos vivos. Causado por una mutación en el gen ARSB, que produce la deficiencia enzimática de arilsulfatasa B ocasionando acumulación de dermatán sulfato en las células. Clínicamente, los pacientes presentan afectación multiorgánica progresiva. En este trabajo presentamos tres pacientes de origen boliviano no emparentados con MPS VI. Adicionalmente revisamos 38 casos de pacientes con MPS VI reportados en literatura.
Palabras clave. Mucopolisacaridosis, Maroteaux-Lamy- ARSB, arilsulfatasa, dermatán sulfato, efecto fundador.
ABSTRACT
The Maroteaux-Lamy syndrome is one of the less common muchopolysaccharidoses subtypes with an approximate incidence of 0.36 to 1.30 per 100,000 live births. MPSVI is caused by the mutation of the ARSB gene, producing the arylsulfatase B enzyme deficiency, that causes cell accumulation of the dermatan sulfate. MPSVI patients progressively develop multiorganic involvement. In this review, we report three unrelated bolivian cases with MPS VI. Additionally we review two series of cases of 38 MPSVI patients reported in literature.
Keywords. Muchopolysaccharidoses, Maroteaux-Lamy-ARSB, arylsulfatase, dermatan sulfate, founding effect.
INTRODUCCIÓN
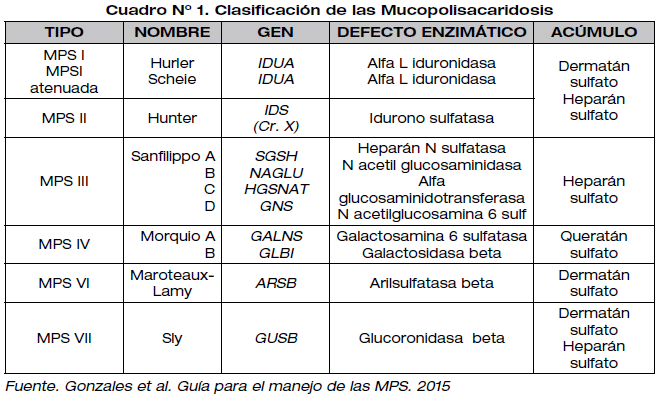
Las mucopolisacaridosis (MPS) se caracterizan por la deficiencia de una enzima lisosomal involucrada en la degradación de azúcares complejos llamados mucopolisacáridos o glucosaminoglucanos (GAGs). Las MPS constituyen enfermedades de afectación multiorgánica de carácter progresivo. Los principales GAGs degradados en los lisosomas son dermatán sulfato, heparán sulfato y queratán sulfato1,2. La incidencia global de las MPS oscila en 1/10000 a 1/22500 aproximadamente, cifra que varía según el tipo de MPS 1,3 . A la fecha se describieron 6 tipos clínicos, que se diferencian por el defecto enzimático, GAG eliminado por orina y el fenotipo que presentan 4. Todas las MPS se transmiten como un carácter autosómico recesivo, con excepción del tipo II, que responde a un patrón de herencia recesivo ligado al cromosoma X 1. El diagnóstico de las MPS se basa en una prueba inicial de detección del tipo de GAG que se excreta por orina o se acumula en el plasma, para posteriormente confirmar la deficiencia enzimática en fibroblastos o leucocitos3. (Ver Cuadro N° 1).
La Mucopolisacaridosis tipo VI (MPS VI; síndrome de Maroteaux-Lamy; OMIM #253200), es uno de los tipos más raros de mucopolisacaridosis. Es causada por mutaciones en el gen Arilsufatasa B (ARSB), localizado en 5q13-5q14, que codifica para la enzima N-acetilgalactosamina -4-sulfatasa (alias arilsulfatasa B (ASB)). La deficiencia enzimática de ASB produce la acumulación de dermatán sulfato en las células 1,5. La incidencia aproximada de la MPS VI es de 0.36 a 1.30 por cada 100,000 nacidos vivos 5. Aparentemente existe una alta tasa de subdiagnóstico, por lo que la incidencia podría ser mucho mayor 5.
La MPS VI clínicamente presenta dos formas: una de progresión rápida y otra de progresión lenta. La primera, tiene un inicio antes de los 2 años, talla baja, facies tosca tipo hurleroide, displasia esquelética con rigidez articular, disostosis múltiple, geno valgo, displasia de cadera, cifoescoliosis; alteraciones oculares (error de refracción, glaucoma, retinopatía, edema del disco óptico y atrofia óptica),
hipoacusia conductiva o neurosensorial, cardiopatía (insuficiencia y/o estenosis mitral o aórtica y cardiomiopatía), afectación pulmonar con infecciones recurrentes, obstrucción de vías áreas, apnea obstructiva del sueño entre otros; estos pacientes fallecen en la segunda o tercera década de la vida, por infecciones, enfermedad cardiaca o complicaciones quirúrgicas. En cambio, el grupo de pacientes con progresión lenta, presenta displasia esquelética más leve y aspecto facial y talla casi normales, lo que retrasa el diagnóstico en la mayor parte de los casos 1,5,6.
Reporte de Casos
Caso clínico 1
Paciente de sexo femenino de 11 años de edad, producto de segunda gestación, sin antecedentes perinatales ni familiares conocidos (condición de abandono). Antecedente de hermano fallecido sin causa aparente en la infancia. La paciente presenta, alteración visual, cardiopatía con lesión valvular y cardiomegalia. Al examen dismorfológico se evidencia talla baja(92cm, p. <3rd), macrocefalia (PC 57cm, p. > 2DE), cráneo alargado, facies triangular, larga y ancha; implantación capilar baja anterior; pabellones auriculares con implantación baja y hélix subdesarrollado bilateral; ojos rasgados, aparente catarata bilateral, cejas espesas y gruesas; puente nasal bajo con narinas anchas; boca grande con paladar alto y macroglosia, agenesia dental con alteración de la dentina y esmalte, hiperplasia gingival; micrognatia; cuello corto y ancho; pectus carinatum, ancho y corto; hernia umbilical; genitales tanner II; escoliosis y cifosis de columna dorsal; miembros superiores con rizomelia y limitación a la extensión articular en codo y hombro bilateralmente; mano en garra, hipoplasia de las falanges distales bilateral; miembros inferiores con rizomelia y geno valgo, con limitación a la extensión en cadera y rodilla; hirsutismo generalizado. No presenta desarrollo puberal. En exámenes complementarios se cuenta con ecocardiografía que reporta válvula aórtica bicúspide, cardiomegalia, lesión mitral y tricúspide leve, hipertensión pulmonar.
El estudio enzimático para mucopolisacaridosis reporta actividad enzimática nula: Aril-sulfatasa "B": 0,0 umol/l/h (valor normal mayor a 5), con lo cual se confirma el diagnóstico de MPS tipo VI.
Caso clínico 2
Paciente de sexo masculino de 11 años de edad, producto de tercera gestación, nacido a término por parto eutócico. Existen antecedentes de familiares lejanos con sospecha de mucopolisacaridosis en ambas líneas parentales. Los padres del paciente proceden del Municipio de Guaqui, provincia Ingavi, departamento de La Paz. Al examen dismorfológico presenta talla baja (109 cm, p. <5th), bajo peso (20 kg, p. <5th). Microcefalia (PC 49.5 cm, p. < 2DE).Turricefalia; facies larga y tosca; pabellones auriculares con cartílagos engrosados; ojos rasgados con cejas anchas y gruesas; puente nasal bajo con narinas anchas; boca grande con paladar ojival y macroglosia, agenesia dental con alteración de la dentina y esmalte, hiperplasia gingival; micrognatia; cuello corto y ancho; pectus carinatum, ancho y corto; hernia umbilical corregida; cifosis dorsal; miembros superiores con rizomelia y limitación de la extensión articular en codo bilateral, mano en garra con dedos cónicos. Miembros inferiores con rizomelia y geno valgo, rigidez y limitación articular en rodilla; hirsutismo generalizado. No presenta desarrollo puberal.
El estudio enzimático para mucopolisacaridosis reporta actividad enzimática nula: Aril-sulfatasa "B": 0,0 umol/l/h (valor normal mayor a 5), con lo cual se confirma el diagnóstico de MPS tipo VI. Presenta test de Berry en orina positivo.
Caso clínico 3
Paciente de sexo femenino de 11 años de edad. Sin antecedentes familiares relevantes. Presentó displasia de caderas, escoliosis dorsolumbar severa, infección respiratoria recurrente, apnea obstructiva del sueño, enfermedad pulmonar restrictiva, problemas tracto respiratorio. Al examen dismorfológico presenta facies típica, macroglosia, hiperplasia gingival, paladar alto, agenesia dental; miopía; cuello corto y ancho; pectus carinatum, ancho y corto; hernia umbilical; miembros superiores con rizomelia y rigidez de las articulaciones de hombro, codos y manos en garra; miembros inferiores con rizomelia, geno valgo y rigidez de articulaciones de cadera y rodilla. Fosita presacra. Hirsutismo generalizado. Las radiografías muestran escoliosis dorsolumbar severa con pico anterior de las vértebras; techos acetabulares planos; costillas con extremo distal adelgazado. No presenta desarrollo puberal. El test de Berry ha sido positivo en orina. El estudio de la enzima Aril-sulfatasa "B", demostró actividad nula.
DISCUSIÓN Y REVISIÓN DE LA LITERATURA
Se revisaron dos series de casos, el primer trabajo reportado por Kiliç et al., 2017 trata de 20 pacientes de origen turco, no emparentados6. La segunda serie de casos reportado por Al-Sanna et al., 2018 trata de 18 pacientes de seis familias, de origen saudí7
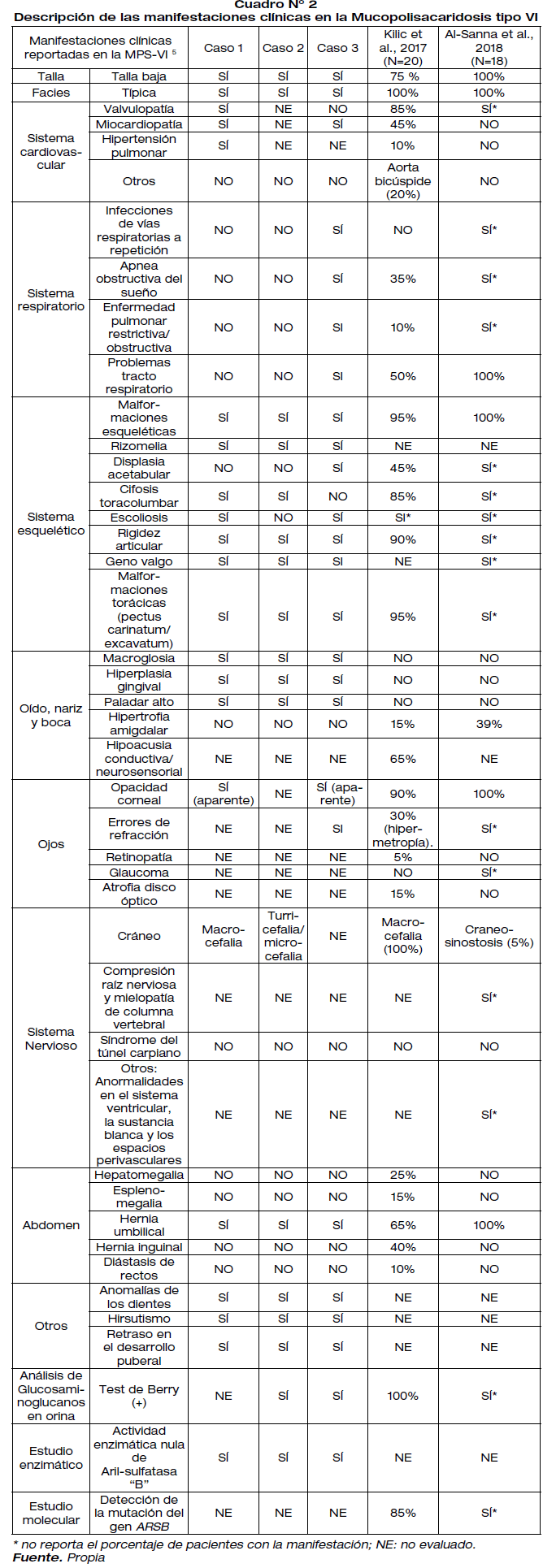
Las manifestaciones clínicas presentes en estos pacientes están detalladas en la Cuadro N° 2. Todos nuestros pacientes corresponden a la forma clínica de progresión rápida. La manifestación fenotípica presente en la revisión de todos los casos revisado fue la facies típica. Coincidentemente, nuestros tres pacientes presentan esta característica clínica.
De las dos series de casos mencionadas, el 70-80% reporta: talla baja, malformaciones esqueléticas y opacidad corneal. Esta última, tiene el 95% de frecuencia de presentación en esta población. De hecho, solo dos de nuestros pacientes presentan opacidad corneal. Sin embargo, las otras dos manifestaciones clínicas se encuentran en nuestros tres casos.
Entre las manifestaciones clínicas más raras reportadas en la serie de casos revisada (< 10%) se encontraron: craneosinostosis, hipertensión pulmonar, retinopatía, enfermedad pulmonar restrictiva/ obstructiva y diástasis de rectos. De estas manifestaciones clínicas, es llamativo encontrar tres características en nuestros pacientes: hipertensión pulmonar en la paciente del caso 1, craneosinostosis en el paciente del caso 2 y enfermedad pulmonar restrictiva en la paciente del caso 3.
La macrocefalia ha sido reportada esporádicamente en la MPS-VI 8"container-title":"Journal of Inherited Metabolic Disease","source":"Pub Med", "abstract" :"BACKGROUND: The mucopolysaccharidoses are multisystem lysosomal storage diseases characterized by extensive skeletal deformities, including skull abnormalities. The objective of this study was to determine the incidence of craniosynostosis in the different mucopolysaccharidosis (MPS. Killic et al., 2017, sugiere que los pacientes con MPS-VI y macrocefalia deben ser evaluados para descartar hidrocefalia 6.
Se ha demostrado la efectividad del tratamiento de remplazo enzimático. Gomes et al., 2018, reportan un efecto positivo en la rigidez articular, alteraciones esqueléticas y la viceromegalia. Aparentemente, no existe cambio en la evolución en el desarrollo motor y la audición 9.
El presente artículo describe por primera vez una serie de 3 pacientes bolivianos, no emparentados, con MPSVI. La caracterización clínica de casos nativos y la comparación con casos presentados en otras poblaciones, ayuda a determinar las particularidades clínicas que pudiesen presentarse asociadas a nuestro grupo poblacional. En nuestra opinión, es importante continuar con el reporte de casos bolivianos y en el futuro identificar la mutación causante, y el haplotipo asociado a la mutación, para descartar la posibilidad de un efecto fundador en nuestra población, como el ya descrito previamente en otras poblaciones.
REFERENCIAS
1. L. Gonzalez Gutierrez & López Marin, L. Guía para el manejo de las Mucopolisacaridosis. (2015).
2. Melbouci, M. et al. Growth impairment in mucopolysaccharidoses. Mol. Genet. Metab. 124, 1–10 (2018).
3. Suarez-Guerrero, J. L. et al. Mucopolisacaridosis: características clínicas, diagnóstico y de manejo. Rev. Chil. Pediatría 295–304 (2016). doi: http://doi.org/10.1016/j.rchipe.2015.10.004
4. Giugliani, R., Harmatz, P. & Wraith, J. E. Management guidelines for mucopolysaccharidosis VI. Pediatrics 120, 405–418 (2007).
5. Harmatz, P. & Shediac, R. Mucopolysaccharidosis VI: pathophysiology, diagnosis and treatment. Front. Biosci. Landmark Ed. 22, 385–406 (2017).
6. Kiliç, M. et al. Genotypic-phenotypic features and enzyme replacement therapy outcome in patients with mucopolysaccharidosis VI from Turkey. Am. J. Med. Genet. A. 173, 2954–2967 (2017).
7. Al-Sannaa, N. A., Al-Abdulwahed, H. Y., Al-Majed, S. I. & Bouholaigah, I. H. The clinical and genetic Spectrum of Maroteaux-Lamy syndrome (Mucopolysaccharidosis VI) in the Eastern Province of Saudi Arabia. J. Community Genet. 9, 65–70 (2018).
8. Oussoren, E. et al. Craniosynostosis affects the majority of mucopolysaccharidosis patients and can contribute to increased intracranial pressure. J. Inherit. Metab. Dis. (2018). doi: http://doi.org/10.1007/s10545-018-0212-110.1007/s10545-018-0212-1
9. Gomes, D. F., Gallo, L. G., Leite, B. F., Silva, R. B. & da Silva, E. N. Clinical effectiveness of enzyme replacement therapy with galsulfase in mucopolysaccharidosis type VI treatment: systematic review. J. Inherit. Metab. Dis. (2018). doi: http://doi.org/10.1007/s10545-018-0212-110.1007/s10545-018-0242-8

