










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica La Paz
versión On-line ISSN 1726-8958
Rev. Méd. La Paz v.19 n.2 La Paz dic. 2013
ARTÍCULO ORIGINAL
ERITROCITOSIS PATOLÓGICA DE ALTURA: CARACTERIZACIÓN BIOLÓGICA, DIAGNÓSTICO Y TRATAMIENTO
HIGH ALTITUDE PATHOLOGICAL ERYTHROCYTOSIS: BIOLOGICAL CHARACTERIZATION, DIAGNOSIS AND TREATMENT
Amaru Ricardo*, Miguez Hortencia, Peñaloza Rosario, Torres Gina, Vera Oscar, Jeaneth Velarde, Nelly Huarachi, Reyna Mamani, Cuevas Heriberto.
* Unidad de Biología Celular, Departamento de Ciencias Funcionales, Facultad de Medicina, Universidad Mayor de San Andrés, La Paz, Bolivia.
Centro de Oncohematología Paolo Belli, La Paz, Bolivia
Correspondencia: Ricardo Amaru MD, PD, ACAD: amaru.ricardo@icloud.com
RECIBIDO: 02-07-2013
ACEPTADO: 07-08-2013
RESUMEN
Introducción. Los pobladores de grandes alturas se adaptaron de modo diferentes, siguiendo rutas distintas con el mismo objetivo de suministro de oxígeno y la supervivencia. En el presente trabajo se caracteriza la Eritrocitosis Patológica de Altura y se demuestra la eficacia de la atorvastatina en el tratamiento.
Material y métodos. Se estudiaron: Sujetos varones como Controles Normales (CN), y pacientes con Eritrocitosis Patológica de Altura (EPA), Eritrocitosis Secundaria (ES) y Policitemia Vera (PV). Se realizaron estudios de laboratorio y de biología molecular. Se realizó estudio clínico de fase 2 con atorvastatina.
Resultados. La EPA presenta: eritropoyetina normal, apoptosis retardada de progenitores eritroides, crecimiento autónomo de BFU-E e hipersensibilidad a la eritropoyetina. La atorvastatina como tratamiento en pacientes con EPA disminuye la concentración de hemoglobina y remite la sintomatología de la hiperviscosidad sanguínea.
Conclusiones. La EPA tiene características propias que la distinguen de otras eritrocitosis patológicas y la atorvastatina se constituye en tratamiento eficaz.
Palabras claves: Eritrocitosis Patológica de Altura, Enfermedad crónica de altura, Estatinas
ABSTRACT
Introduction. The residents of high altitude get use to it in different ways, following the same purpose in order to supply oxygen and survival. In the present paper, we characterize the High Altitude Pathological Erythrocytosis disease and demonstrate the effectiveness of atorvastatin in its treatment.
Material and methods. We studied male subjects as normal controls (CN), and patients with High Altitude Pathological Erythrocytosis (EPA), Secondary Erythrocytosis (ES) and Polycythemia Vera (PV). Laboratory and molecular biology studies were conducted. We developed in the study of 2nd phase with atorvastatin.
Results. The EPA presents: normal erythropoietin, delayed erythroid progenitors apoptosis, autonomous growth of BFU-E and hypersensitivity to erythropoietin. The atorvastatin in patient with EPA, decreased hemoglobin concentration and eliminates the symptomatology of blood hyperviscosity
Conclusions. The EPA has characteristics that distinguish it from other pathologic erythrocytosis and atorvastatin becomes an effective treatment.
Keywords: High Altitude Pathological Erythrocytosis, Chronic Mountain Sickness, Statins.
INTRODUCCIÓN
La tierra, desde su formación, ha estado marcada por acontecimientos importantes, como la formación de la montaña Himalaya (Tibet) entre 245 a 65 millones de años y la formación de la Cordillera de Los Andes entre 138 a 65 millones de años1,2. Las poblaciones nativas del Tibet y Los Andes colonizaron las montañas de Tibet y Los Andes desde hace 25.000 y 11.000 años atrás, respectivamente3. La baja presión de oxígeno en grandes alturas en las que vivieron ambas poblaciones permitió la selección natural y adaptación genética4 de modo diferentes como resultado de procesos evolutivos y tiempo de exposición distintos. Los Tibetanos montañeses evolucionaron hacia una concentración de hemoglobina similar a los habitantes del nivel del mar, gracias a la adaptación y selección positiva de un grupo de genes involucrados en la eritropoyesis; mientras que los Andinos evolucionaron a hemoglobina elevada en relación a los habitantes del nivel del mar. Además, los nativos Tibetanos y Andinos presentan una baja concentración de hemoglobina en relación a los nativos de tierras bajas que migraron recientemente a grandes alturas5,6,7,8,9,10.
La eritrocitosis es el aumento de la masa de eritrocitos por encima de los parámetros normales. Las principales eritrocitosis patológicas son: Policitemia Vera (PV), Eritrocitosis Secundaria (ES), Eritrocitosis Familiar (EF) e Eritrocitosis Patológica de Altura (EPA). La PV es una enfermedad oncohematológica clonal11 asociada a leucocitosis, trombocitosis12, crecimiento autónomo de colonias eritroides (BFU-E), eritropoyetina sérica baja13,14 y la mutación de gen JAK2 V617F15,16,17,18 propio de las enfermedades mieloprolierativas19,20,21,22,23. La ES representa la consecuencia de patologías asociadas a aumento de la eritropoyetina sérica como las patologías cardiopulmonares 24,25,26,27. La EF es genético y se caracteriza por la mutación de genes involucrados en la eritropoyesis 28,29. La EPA es la manifestación hematológica de la enfermedad crónica de altura, presente en nativos o residentes en alturas por encima de 2500 msnm 30,31,32,33 probablemente de etiología multifactorial34,35,36,37,38 y la inadecuada adaptación a las grandes alturas; la incidencia en la ciudad de La Paz a 3.600 msnm es de 5 a 7%39,40. Para el tratamiento de la EPA se ha recurrido a medidas terapéuticas como la flebotomía41, el uso de Medroxiprogesterona42, Acetazolamida43, Almitrina44, teofilina45,46 y enalapril47. Por diferentes factores y por presencia de eventos adversos actualmente no son utilizados como medicamentos de primera línea.
MATERIAL Y MÉTODOS
Pacientes
Para el estudio clínico se estudiaron 15 sujetos varones como Controles Normales (CN), 17 varones con EPA, 51 varones con ES y 8 pacientes con PV. Para estudio biomolecular se estudiaron 10CN, 10EPA, 10ES y 5PV. Para el estudio clínico con atorvastatina se estudiaron 22 EPA con radicatoria en las ciudades de La Paz y El Alto a 3600 y 4000 msnm respectivamente. Las muestras de aspirado de médula ósea y sangre venosa periférica fueron tomadas previo consentimiento informado, y los datos clínicos se obtuvieron de la historia clínica. El seguimiento de los pacientes fue realizado mensualmente por consultorio externo. El diagnóstico de eventos trombóticos fue confirmado con Eco doppler y Tomografia axial computarizada.
Análisis de laboratorio
La concentración de hemoglobina, hematocrito, leucocitos y plaquetas fueron realizados en sangre venosa periférica obtenida en tubos con EDTA (Becton Dickinson, USA) y procesados en un contador hematológico automático (Micro 60, USA). La hemoglobina fetal se determinó por método fotocolorímétrico, con el Kit Emoglobina Fetale Kit (Globe Diagnostic, Italy) en un espectrofotómetro (Unico 1200, USA) y la methemoglobina fue estudiada con el método de Evelyn y Malloy. Los reticulocitos se evaluaron por microscopia mediante tinción azul brillante de cresil. La dosificación de eritropoyetina sérica, se realizó en sangre venosa periférica obtenida en tubos Vacutainer SST II Advance (Becton Dickinson, Plymouth, UK) con el método de ELISA, utilizando Kit comercial (R&D System, USA) y un lector Stat Fax 2100 (Awarenes Inc. Technologies, USA). La gasometría arterial se realizó en gasómetro automatizado (Critical Care Xpress, Nova Biomedical).
Aislamiento de células progenitoras hematopoyéticas (CPH)
Las CPH de la médula ósea fueron separadas con centrifugación por gradiente de densidad Histopaque 1077 (Sigma-Aldrich, USA), luego lavadas con tampón de lisis y RPMI1640 con suero fetal bovino 2%, e identificadas por citometría de flujo con anti CD34, y la viabilidad se estudió con Azul Tripan.
Análisis de Inmunofenotipo por Citometría de Flujo
Las muestras fueron tomadas en tubos Trucount de poliestireno (Becton Dickinson, USA) y marcados con anticuerpos CD34, CD45 y Gly-A (Becton Dickinson, USA). La evaluación se efectuó en Citómetro de flujo FASCAN (Becton Dikinson, USA) y fueron evaluados con los programas Multiset y Cell Quest Pro (Becton Dickinson-USA).
Análisis de la mutación del gen Jak-2 V617F
El DNA de células mononucleares de la médula ósea fue extraído con método fenol/cloroformo. Se utilizó 100 ng de DNA, 50 pM de los oligos JAK2R (5'CTGAATAGTCCTACAGTGTTTTCA GTTTCA3'), JAK2F (5'AGCATTTGGTTT TAAATTATGGAGTATATT3') y JAK2FWT (5'ATCTATAGTCATGCTGAAAGTAGGAG AAAG3'). El annealing se realizó a 59°C por 35 ciclos. El producto del PCR fue separado en gel de agarosa al 2%. La banda de 364 bp representa el exon 12 del gen Jak2 y la banda 203 bp la mutación Jak2 V617F.
Cultivo de CPH en medio semisólido
Se cultivó 2x105cel/ml de CPH en cajas petri de 33 mm con grilla de acuerdo al siguiente protocolo: 1 ml de MetIlcelulosa (Metho Cult H4230 Without Epo, stemCell Technologies, Canada), penicilina 250 UI/ml, estreptomicina 50ug/ml, simvastatina 300ng/ml y Epo 2UI/ml (los controles sin Epo ni simvastatina). El cultivo fue realizado a 37°C y 5% CO2. La lectura de las colonias BFU-E se realizó de acuerdo a criterios estandarizados a los 7 y 14 días48.
Cultivo de CPH en medio líquido
Los cultivos de CPH se realizaron en placas de poliestireno de 12 y 24 pocillos (Cellstar greiner Bio-one USA) en RPMI 1640 con suero fetal bovino 10%, L-glutamina 2mM, estreptomicina 50Ug/ml, penicilina 50UI/ml, neomicina 1000Ug/ml, rHuEpo 3 UI/ml (Hemax, SIDUS) y simvastatina 10UM (Merk Sharp Dohm, France). Se cultivaron 1x106 cel/ ml a 37°C al 5% de CO2 durante 5 días en una estufa de CO2 (Binder GMBH, Germany).
Dosis respuesta de CPH a la eritropoyetina
Se cultivaron 2x105cel/ml en metilcelulosa (MethoCult 4531 medium, StemCell Technologies Inc, Vancouver, Canada) con penicilina 250 UI/ml, estreptomicina 50ug/ml, en cajas Petri de 35 mm en presencia de eritropoyetina a las siguientes concentraciones: 0; 0.03; 0.06; 0,125; 0.250 y 2 U/ml. El cultivo fue realizado a 37°C ,5% de CO2 hasta el día 14. La lectura de BFU-E se realizó de acuerdo a criterios estandarizados.
Determinación de apoptosis por citometría de flujo.
La apoptosis celular se determinó mediante el Kit de Apoptosis (Becton Dickinson, Pharmigen, USA) con anti-CD235a, Anexina V-PE y 7AAD. La evaluación se realizó por citometría de flujo con FACScan (Becton Dickinson, USA) y el software Cell Quest Pro 4.0 (Becton Dickinson, USA).
Determinación de apoptosis por Cultivo en medio semisólido
Se cultivó 2x105cel/ml en cajas petri de 33 mm con grilla y en 1 ml de MetIlcelulosa (Metho Cult H4230 Without Epo, stemCell Technologies, Canada) suplementado con penicilina 250 UI/ml, estreptomicina 50ug/ml, simvastatina 300ng/ml y Epo 2UI/ml. El cultivo fue realizado a 37°C, 5% de CO2. La lectura de las colonias BFU-E se realizó a los 7,14 y 21 días 48.
Determinación de apoptosis por DNA laedder
Se cultivaron 5x106 cel/ml de células mononucleares en 700ml RPMI-1640, 300 ml suero fetal bovino, L-Glutamina 2nM, Penicilina 250UI, estreptomicina 50mg y rHEpo 2UI/ ml. El cultivo se realizó en placas CCW (Corning Cell Well) a 37°C, 5% de CO2. Fueron cultivadas durante 1, 2, 7 y 14 días, luego lavadas con PBS y lisados en solución guanidio y proteínasa K. Posteriormente fueron incubados a 58°C por 60 min y separados por electroforésis en gel de agarosa al 2%. El patrón de DNA fue visualizado y fotografiado bajo luz ultravioleta.
Tratamiento de pacientes con Eritrocitosis Patológica de Altura
Veinti y dos pacientes con EPA asociados a hipercolesterolemia recibieron 20 mg de Atorvastatina por via oral a horas 21:00 cada día. La duración del tratamiento fue por más de un año y la evaluación se realizó a los 12 meses de estudio. Se determinó suspender el tratamiento en pressencia de eventos adversos, intolerancia a la atorvastatina y/o solicitud del paciente. Los tratamientos concomitantes prohibidos fueron la Eritropoyetina, Estrógenos, Andrógenos y Corticoides. Previo inicio de tratamiento se realizó sangría de 450 ml semanales hasta alcanzar hemoglobina inferior a 18 g/dl. Se tomaron las siguientes medidas para valorar el cumplimiento del tratamiento: visita médica mensual, entrevista, confirmación y llamadas telefónicas. Se tomaron los siguientes criterios para la valoración de respuesta a la Atorvastatina:
Respuesta Completa: hemoglobina ≤ 17.5 g/dl, remisión de signosintomatología y sin requerimiento de flebotomías.
Respuesta Parcial: hemoglobina 17.5 a 19 g/dl, presencia de uno o más signosintomatologías y con requerimiento de sangrías.
Respuesta Ausente: hemoglobina superior a 19 g/dl, presencia de uno o más y con requerimiento de sangrias.
RESULTADOS
Características clínicas
Las características clínicas de los pacientes estudiados esta detallado en el Cuadro N°1.
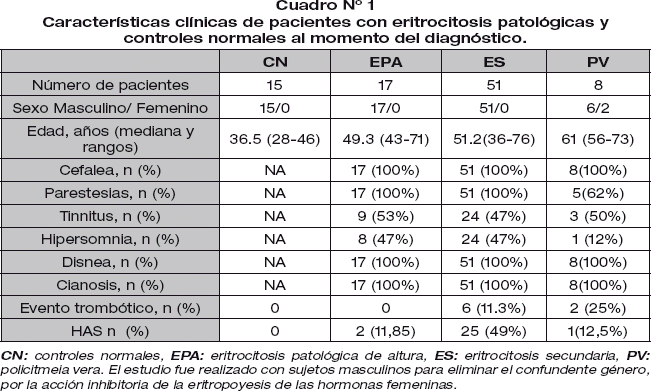
El motivo de consulta de los pacientes con eritrocitosis patológica (EPA, ES, PV) fue el síndrome de hiperviscosidad sanguínea caracterizado por: cefalea, parestesias, tinnitus, hipersomnia y disnea, además de la cianosis distal. Los pacientes con ES presentaron signos pulmonares evidentes de patología orgánica y los pacientes con PV presentaron leucocitosis, trombocitosis y esplenomegalia.
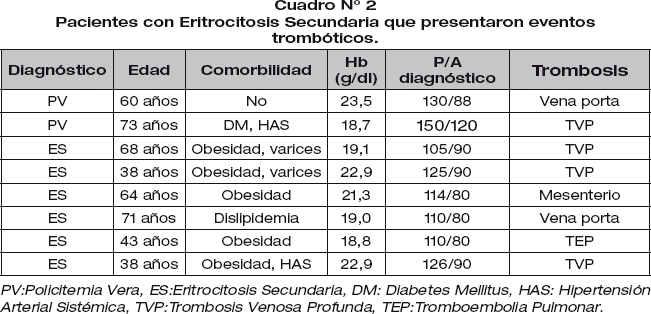
La historia de evento trombótico fue del 11.3% en ES y 25% en PV. El evento trombótico en ES estuvo asociado a obesidad y dislipidemia (Cuadro N° 2).
Al diagnóstico presentaron Hipertensión Arterial sistémica (HAS) 11.8% de pacientes con EPA, 49% con ES y 12% con PV. Posterior a sangría hasta alcanzar valores menores de 18 g/dl, la HAS se normalizó en pacientes con EPA y PV; mientras el 11.5% de pacientes con ES continuaban con HAS, estos últimos estaban asociados a obesidad.
Características laboratoriales
Las características laboratoriales de los pacientes estudiados están detallados en el cuadro N° 3.
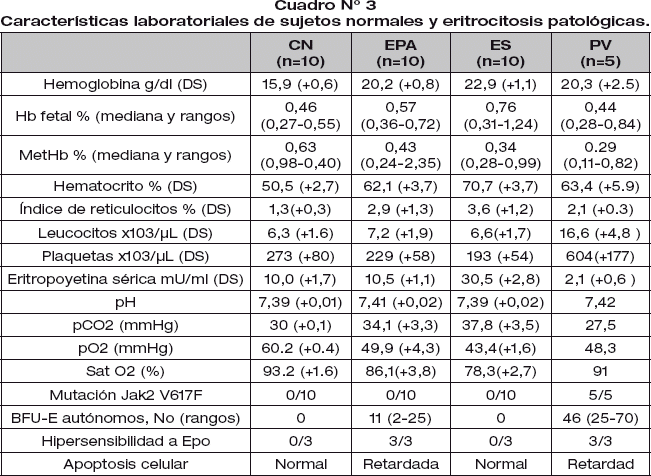
Los pacientes con EPA tienen la hemoglobina y hematocrito estadísticamente diferente de los pacientes con ES, PV y CN; mientras que, el índice de reticulocitos son similares a los pacientes con ES y PV y estadísticamente diferentes de los CN. La concentración de hemoglobina fetal y metahemoglobina en pacientes con EPA, ES y PV son normales.
La cantidad de leucocitos y plaquetas son similares en CN, EPA y ES; mientras que los pacientes con PV presenta leucocitosis y trombocitosis.
La concentración de eritropoyetina sérica es normal en el grupo EPA, elevada en ES y disminuida en PV.
El pH de los cuatro grupos (SN, EPA, ES y PV) son normales; pero, la pCO2, pO2 y la saturación de O2 de los pacientes con EPA son estadísticamente diferentes de los pacientes con ES y CN.
La mutación de JAK2 V617F esta presente en todo los pacientes con PV y ausente en EPA, ES y CN (fig 1).

Electroforesis de producto de PCR en gel de agarosa al 2%. Las muestras del control, Eritrocitosis Patológica de Altura y Eritrocitosis secundaria no presentan mutación del gen JAK2 V617F. La muestra de la policitemia presenta mutación del gen JAK2 V617F.
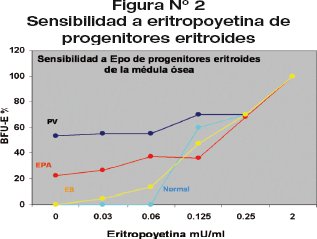
El crecimiento autónomo de colonias eritrocitarias BFU-E, sin presencia de eritropoyetina, estuvo presente en EPA; pero, no en la misma intensidad y cantidad que los pacientes con PV, cuya característica es el BFU-E autónomo.
Los progenitores eritroides de pacientes con EPA en medio de cultivo celular metilcelulosa presentan hipersensibilidad a bajas concentraciones de eritropoyetina (Fig. 2).
Crecimiento de BFU-E en ausencia de eritropoyetina y concentraciones bajas de eritropoyetina de progenitores eritroides en medio de metilcelulosa.
Las pruebas de apoptosis y necrosis realizados en cultivos celulares en medio líquido, medio semisólido y por degradación de DNA demuestran que los progenitores eritroides de pacientes con EPA presentan apoptosis retardada en relación a los progenitores eritroides de sujetos normales (fig 3).
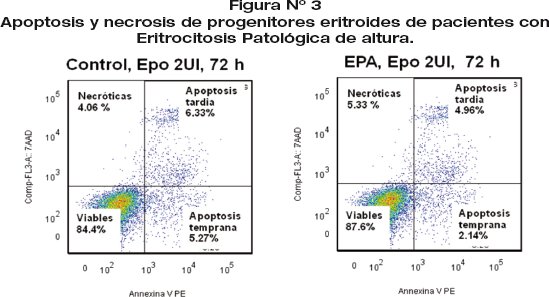
Dot Blot de apoptosis y necrosis de progenitores eritroides de sujetos normales y pacientes con EPA. La viabilidad celular se determinó por la expresión de Anexina-V y 7AAD de la siguiente manera: células viables: Anexina V negativo y 7AAD negativo, células apoptóticas tempranas: Anexina V positivo y 7AAD negativo, células apoptóticas tardías: Anexina V positivo y 7AAD positivo, células necróticas: Anexina V negativo y 7AAD positivo.
Acción "in vitro" de las estatinas sobre la eritropoyesis.
Las estatinas, en modelos celulares, inducen apoptosis de progenitores eritroides e inhiben la proliferación y diferenciación de Unidades Formadoras de Colonias Eritrocitarias (BFU-E) de pacientes con EPA, ES,PVy CN (fig 4).
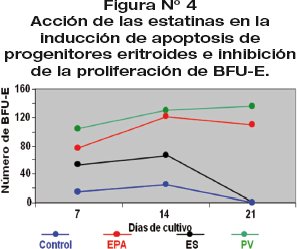
El estudio de la apoptosis en medio semisólido (metilcelulosa) fue realizado por citometria de flujo.
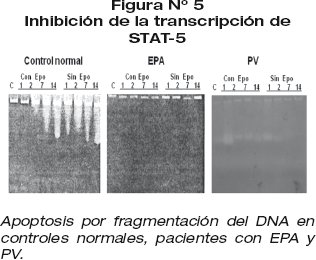
La acción de las estatinas en vitro, en medio de cultivo celular líquido inhibe el factor de trascripción STAT-5 (fig 5).
Acción "in vivo" de las estatinas sobre la eritropoyesis.
Los pacientes con EPA tratados con 20 mg día de atorvastatina, 64% alcanzaron remisión completa, 27% remisión parcial y 9% no respondió al tratamiento (cuadro N° 4).
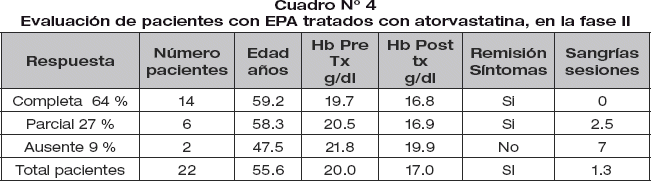
El estudio se realizó en 10 pacientes con EPA asociados a hipercolesterolemia, recibieron 20 mg de Atorvastatina por via oral cada 24 horas. La evaluación se realizó a los 12 meses. Previo inicio de tratamiento se realizó sangría de 450 ml semanales hasta alcanzar hemoglobina inferior a 18 g/dl. Se siguieron 22 pacientes con eritrocitosis patológica de altura que recibieron atorvastatina 20 mg VO día durante 12 meses; se observaron datos hematológicos y sintomatología del síndrome de hiperviscosidad.
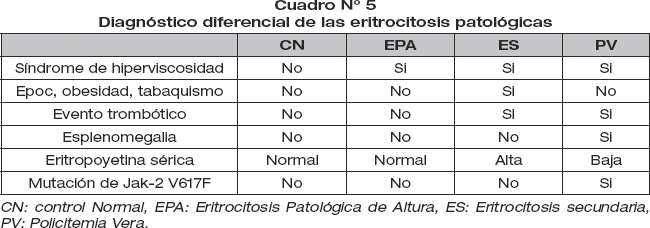
Diagnostico diferencial de las eritrocitosis patológicas.
Con los datos obtenidosse es posible el diagnóstico diferencial entre las eritrocitosis patológicas más frecuentes (cuadro N° 5).
DISCUSIÓN
La Eritrocitosis Patológica de Altura (EPA) es la manifestación hematológica de la Enfermedad Crónica de Altura (enfermedad de Monge), caracterizado por el aumento de número de eritrocitos, hemoglobinas y hematocrito, que se manifiesta clínicamente por el síndrome de hiperviscosidad sanguínea y cianosis. Es un cuadro clínico por adaptación inadecuada a grandes alturas y probablemente por falta de adaptación de un grupo de genes comprometidos en la eritropoyesis.
Los valores de la concentración de hemoglobina en condiciones normales y patológicas dependen de la edad, altura de residencia y sexo49,50,51; los valores normales para los habitantes adultos de las ciudades de La Paz y El Alto son: de 14 a 17 g/dl para mujeres y de 15 a 18 g/ dl para varones. En el presente estudio se consideró eritrocitosis hemoglobina mayor a 18 g/dl para las mujeres y mayor a 19 g/dl para los varones; por encima de estos valores más del 80% de mujeres y varones presentan sintomatología de hiperviscosidad sanguínea. Pero es importante mencionar que existen pacientes de ambos sexos que toleran sin sintomatología hemoglobinas elevadas y a la inversa hay pacientes, sobre todo mujeres, que presentan sintomatología de hiperviscosidad a valores inferiores al del cut off.
La incidencia de la EPA no está determinada, por falta de criterios de diagnóstico diferencial entre la EPA, ES y la PV. Sin embargo varios autores han citado la incidencia de 5 al 10% 51,52,53 en regiones por encima de 2500 msnm; pero, probablemente la EPA sea menos frecuente. La ES es la mas frecuentes de todas las eritrocitosis patológicas, que en la altura se manifiesta con más sensibilidad; por ejemplo pacientes con daño pulmonar leve, en la altura presentan eritrocitosis, mientras que este mismo paciente a nivel del mar probablemente no presente la eritrocitosis. Se presenta generalmente en varones en edad adulta y mujeres post menopaúsicas, que fue adecuadamente descrito desde el punto de vista fisiopatológico 54,55,56. De acuerdo a nuestras primeras observaciones, de todas la eritrocitosis patológicas, 90% son ES, 9% EPA y 1% corresponde a PV.
La EPA se manifiesta por la sintomatología del síndrome de hiperviscosidad sanguínea caracterizada por cefalea, disnea, parestesias, hipersomnia y tinnitus57,58, asociado a cianosis periférica. En nuestro estudio todos los pacientes con eritrocitosis patológica (EPA, ES, PV) presentan síndrome de hiperviscosidad sanguínea y cianosis; cuando la eritrocitosis se asocia a hipertensión arterial sistémica secundaria a hipervolemia se observa epistaxis. Los pacientes con EPA seguidos por más de 3 años, no presentaron eventos trombóticos, probablemente la eritrocitosis aislada no es un factor de riesgo para eventos tromboticos; es raro los eventos tromboticos en eritrocitosis no neoplásicos59,60 y en eritrocitosis inducida en modelos animales61. La ES asociada a síndrome metabólico tienen probabilidad alta de presentar evento trombótico y la PV tiene una alta probabilidad de presentar evento trombótico por la presencia de factores procoagulantes propios de enfermedad neoplásica62,63,64,65.
La hemoglobina y hematocrito de los pacientes con EPA, ES y PV están elevadas y los valores de EPA son estadísticamente diferentes de ES y PV, probablemente porque la etiología de cada uno de estas entidades clínicas son diferentes 66,67,68. El índice de reticulocitos es similar en la ES, EPA y PV como manifestación de patologías que tienen en común el aumento de la eritropoyesis. La EPA y la ES no presentan alteraciones en otras líneas hematopoyéticas, como ocurre en la PV que está asociada a leucocitosis y trombocitosis69,70; lo que hace suponer que la EPA y ES tienen alteraciones en la línea eritroide exclusivamente.
La concentración de eritropoyetina sérica de los pacientes con EPA está dentro de los rangos normales, es estadísticamente diferente de los pacientes con ES donde se halla elevada 71,72 y PV cuya concentración esta muy disminuida67,70.
La EPA presenta hipercapnia e hipoxemia leve que puede ser manifestación del aumento de la viscosidad sanguínea, como sucede en los pacientes con PV 72,73. La EPA y ES no presentan la mutación de JAK-2 V617F, y es positiva en todo los casos de PV 74,75. El crecimiento autónomo de las BFU-E es característica de la PV como manifestación de enfermedad clonal 76,77,78,79, mientras que el crecimiento autónomo en pacientes con EPA podría entenderse como hipersensibilidad a la eritropoyetina por parte de los progenitores eritroides. La EPA presenta apoptosis retardada, probablemente el gen Bcl-xL, responsable de la acción antiapoptotica de la serie eritroide, este involucrado80,81,82.
El diagnóstico diferencial de la EPA con otras eritrocitosis patológica se caracteriza por la diferencia de concentración de Hemoglobina, leucocitosis, trombocitosis, concentración de eritropoyetina, ausencia de mutación del JAK-2V617F, crecimiento autónomo de los progenitores eritroides y la esplenomegalia (ver cuadro 5).
La etiología de la EPA pareciera ser de naturaleza genética. Los genes involucrados en la eritropoyesis en la altura estarían en vías de adaptación. Los principales genes comprometidos en la adaptación son: HIF-2α, Citocromo P450, EDNRA, ANGPTL4, CAMK2D, EGLN1, HMOX2, CYP17A1, PPARA y PTEN; si este grupo de genes se adaptan a la altura, la concentración de hemoglobina de las generaciones desciende; si no se adapta a la altura, la concentración de hemoglobina de las generaciones aumenta. Pareciera que el gen más importante en la adaptación a grades alturas es el HIF-2α (Factor Inducible a la Hipoxia 2 alfa) que desempeña un papel importante en la regulación de la eritropoyesis, y que junto al gen EGLN1 (un regulador del HIF), y PPARA (un objetivo transcripcional del HIF) están fuertemente asociados con la disminución de la concentración de la hemoglobina en Tibetano montañeses. Los portadores del alelo «Tíbet» de los HIF-2α son capaces de mantener la suficiente oxigenación de los tejidos a gran altura sin la necesidad de aumento de eritrocitos. Las poblaciones andinas, probablemente a causa de su corta historia de residencia a grandes alturas no han desarrollado un mecanismo similar para disminuir la eritropoyesis; sin embargo las poblaciones andinas presentan eritrocitosis como mecanismo de compensación a grandes alturas83,84,85, probablemente por la hipersensibilidad a eritropoyetina y apoptosis retardada de los progenitores eritroides. La hipersensibilidad también se observa en pacientes con eritrocitosis familiar caracterizados por la mutaciones en el gen del EpoR86,87,88 en progenitores eritroides de pacientes con eritrocitosis post trasplante renal y en pacientes con policitemia vera. En pacientes con ES la sensibilidad a eritropoyetina es normal89,90.
La EPA presenta como principales complicaciones la hipertensión arterial sistémica secundaria a hipervolemia y la hipertensión arterial pulmonar. El evento trombótico no constituye una complicación en pacientes con EPA, sin embargo en pacientes con ES asociado a síndrome metabólico (ES/SM) presentan como principales complicaciones el evento trombótico, hipertensión arterial sistémica e hipertensión arterial pulmonar. Además en un paciente con ES/SM se observó insuficiencia cardiaca congestiva que fue resuelto con sangría. Otra complicación observada fue el síndrome convulsivo en una paciente con Hb por encima de 24 g/dl. Las alteraciones de coagulación y hemostasia están actualmente en estudio 91,92,93.
El tratamiento de la EPA con atorvastatina, en las fases I y II, mostraron significativa reducción de la concentración de hemoglobina y la remisión de la sintomatología de la hiperviscosidad sanguínea. El 60 % de los pacientes tratados alcanzaron respuesta completa con una calidad de vida buena y performance status 0. Los pacientes con Respuesta Parcial, a pesar de tener hemoglobinas por encima de valores normales, retornaron a sus actividades cotidianas.
Para el tratamiento de pacientes con EPA (también válido para pacientes con ES) se propone el siguiente protocolo:
a) Sangrías semanales hasta alcanzar valores menores de 18 g/dl de Hemoglobina. Sangrías de 450 ml en pacientes hemodinamicamente estables, minisangrias de 250 ml en pacientes con presiones arteriales bajas y pacientes mayores de 60 años, y microsangrias de 100 ml en pacientes con insuficiencia cardiaca y mayores de 70 años. Las reposiciones están contraindicadas en pacientes con hipertensión arterial sistémica; la reposición con solución fisiológica de 500 ml es solo recomendado para pacientes con presión arterial baja que presentan hipotensión posterior a sangrías.
b) Atorvastatina 20 mg VO día, horas 21:00. Durante el tratamiento monitorizar la CPK (Creatinfosfokinasa en sangre) y los efectos adversos de las estatinas.
c) Sangrias adicionales en los siguientes casos:
Presencia desíndrome de hiperviscosidad sanguínea
Hematocrito superior a 60% o hemoglobina superior a 19 g/dl.
Las estatinas son inhibidores de la hidroximetil glutaril Co enzima A reductasa, cuyo objetivo es la inhibición de la síntesis endógena del Colesterol; pero, además tiene acciones pleiotropicas que intervienen en la proliferación, diferenciación y apoptosis de las cé lulas94,95,96,97,98,99,100,101,102,103,104,105. Esta propiedad propia de las atorvastatinas inhibe la eritropoyesis seguida de la disminución de la concentración de la hemoglobina, remisión de la sintomatología de la hiperviscosidad sanguínea, normalización de la presión arterial sistémica y probablemente reduce la hipertensión arterial pulmonar (datos no publicados).
REFERENCIAS
1. Joel Cracraft.«The Scientific Response to Creationism ». Department of Astronomy, University of Illinois. 1982. . es.wikipedia.org/wiki/Historia_de_la_Tierra. [ Links ]
2. Futuyma, Douglas J. Evolution. Sunderland, Massachusetts: Sinuer Associates, 2005. ISBN0-87893-187-2. es.wikipedia.org/wiki/Historia_de_la_vida [ Links ]
3. Storz J. Genes for High Altitudes. Science 2010;329:40-41 [ Links ]
4. Moore LG. Human genetic adaptation to high altitude. High Alt Med Biol. 2001 ;2:257-279. [ Links ]
5. Tatum S. Simonson. Genetic Evidence for High-Altitude Adaptation in Tibet. Science 2010; 329:72-75
6. Mark Aldenderfer. Peopling the Tibetan Plateau: Insights from Archaeology. High Altitude Medicine & Biology. 2011 ;12:141-147.
7. Simonson TS, McClain DA, Jorde LB, Prchal JT. Genetic determinants of Tibetan high-altitude adaptation. Hum Genet. 2012;131:527-33. [ Links ]
8. Moore LG. Human genetic adaptation to high altitude. High Alt Med Biol. 2001:257-79.
9. Moore LG, Armaza F, Villena M, Vargas E. Comparative aspects of high-altitude adaptation in human populations. Adv Exp Med Biol. 2000;475:45-62. [ Links ]
10. Aparicio Octavio. Texto de Medicina de Altura. GMC Artes Gráficas, 2008.77-79. [ Links ]
11. Adamson JW: Polycythemia vera: Stem-cell and probable clonal origin of the disease. N Engl J Med 1976;295:913. [ Links ]
12. Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood 2002, 100:4272-4290. [ Links ]
13. Prchal JF. The in vitro response of normal and abnormal stem cell lines to erythropoietin. J Clin Invest 1978;61:1044. [ Links ]
14. Eaves CJ: Erythropoietin dose-response curves for three classes of erythroid progenitors. Blood 1978;52:1196. [ Links ]
15. Jones A. Widespread occurrence of the JAK2 V617F mutation. Blood 106: 2162-2168.
16. Levine RL: Activating mutation in the tyrosine kinase JAK2 in polycythemia vera. Cancer Cell 2005;4:387. [ Links ]
17. Kralovics R: A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005;352:1779. [ Links ]
18. Baxter EJ. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054. [ Links ]
19. James C. A unique clonal JAK2 mutation leading to constitutive signalling causes PV. Nature 434:1144-1148.
20. Zaleskas V. Molecular Pathogenesis and Therapy of Polycythemia Induced in Mice by JAK2. PLoS ONE.2006.
21. Finazzi G. Expertise-based management in essential thrombocythemia and PV. Cancer J. 2007;13:372-376. [ Links ]
22. Finazzi G. The treatment of polycythaemia vera: an update in the JAK2 era. Intern Emerg Med. 2007;2:13-18. [ Links ]
23. Bellucci S. The role ofJAK2 V617F mutation. Semin Thromb Hemost. 2006;32:381-398. [ Links ]
24. Flenley DC: Chronic obstructive pulmonary disease. Dis Mon 1988;34:537 [ Links ]
25. Limthongkul S: Chronic obstructive pulmonary disease at Chulalongkorn Hospital. J Med Assoc Thai 1991;74:639 [ Links ]
26. Guidet B. Polycythemia in chronic obstructive pulmonary disease. Chest 1987;92:867-870. [ Links ]
27. Villanueva-Gimenoa MM, Vicario-Bermúdeza JM. Eritrocitosis secundaria a la producción inapropiada de eritropoyetina por un carcinoma de células renales. Semergen 2012;454:1-3. [ Links ]
28. Prchal J, Sokol L. Familiar polycythemia. Sciencie 1995;1831-1832. [ Links ]
29. Melanie J Percy. Familial erythrocytosis arising from a gain-of-function mutation in the HIF2A gene of the oxygen sensing pathway. Ulster Med J 2008; 77 (2) 86-88. [ Links ]
30. Gonzales GF. Contribución Peruana a la hematología en poblaciones nativas de altura. Acta Andina 1998; 7: 105. [ Links ]
31. Leon-Velarde F: Hematological parameters in high altitude residents living. High Alt Med Biol 2000;1:97. [ Links ]
32. León-Velarde F. Consensus statement on chronic and subacute high altitude diseases. High Alt Med Biol. 2005;6:147-57. [ Links ]
33. Amaru R. Novedoso tratamiento farmacológico de la eritrocitosis patológica de altura. Revista médica, 2004;10:7.
34. Moore LG. Does chronic mountain sickness have perinatal origins?. Respir Physiol Neurobiol. 2007;158:180-189. [ Links ]
35. Spicuzza L. Sleep-related hypoxaemia and excessive erythrocytosis in Andean natives. Eur Respir J. 2004;23:41. [ Links ]
36. Kryger M. Excessive polycythemia ofhigh altitude. Am Rev Respir Dis. 1978;118: 659. [ Links ]
37. Amaru Ricardo. Alteración en el patrón de apoptosis de células precursoras de eritrocitos. Cuaderno 2000;46:31. [ Links ]
38. León-Velarde F. Respiratory control in residents at high altitude. High Alt Med Biol. 2006; 7:125-137. [ Links ]
39. Spielvogel Hilde, Paz Zamora M. Tufts D. Comunicación rápida IBBA. 1988.
40. Vargas E. Excessive erythocythemia and chronic mountain, studies from the IBBA. Project Alfa. 2005.74.1985. [ Links ]
41. Manier G. Pulmonary gas exchange in Andean natives with excessive polycythemia. J Appl Physiol.1988;65:2107-2117.
42. Kryger M. Imapired oxygenation during sleep in excessive polycythemia of high altitude. Sleep. 1978;1:3-17. [ Links ]
43. Richalet JP. Azetozolamide: a treatment for chronic mountain sickness. High alt Med Biol. 2004;5:258. [ Links ]
44. Villena M. Double-blind study on the action of almitrine in patients with polycythemia of high altitude. Bull Eur physiopathol Respir. 1985;21:165-170. [ Links ]
45. Oren R, Berri M, Hubert A, Matzner Y. Effect of Theophylline on erythrocytosis in chronic obstructive pulmonary disease. Archives of Internal Medicine 1997;157:1474-1478. [ Links ]
46. Romero R, Lens M. Tratamiento de la poliglobulia post trasplante con teofilina. Nefrologia 1992;12:174. [ Links ]
47. Plata R. Angiotensin-converting-enzyme inhibition therapy in altitude polycythaemia. Lancet. 2002;359:663-666. [ Links ]
48. Montagna C, Eridani S. In vitro sensitivity of human erythroid progenitors to hemopoietic rowth factors. Haematologica 1994;79:311-318. [ Links ]
49. Vargas E, Spielvogel H. High altitude medicine & biology. 2006;7:2. [ Links ]
50. Brito J. Chronic intermittent hypoxia at high altitude exposure for over 12 years. High Alt Med Biol. 2007;8:236-244. [ Links ]
51. Spielvogel H, Paz Zamora M, Daigh A Tuts, Jere D, Haas L. Sobre la incidencia de la eritrocitosis en la población masculina de La Paz. IBBA 1988;2:17-27. [ Links ]
52. Vargas E, Villena M. Factores predominantes en la etiopatogenia de la enfermedad de Monge (EPA) en La Paz Bolivia.IBBA:263-282 [ Links ]
53. Galarza M. Rodrigo G. Comunicación de la segunda hemoglobina mutada en Bolivia, Hb Illimani. Cuad Hosp Clin 1991;37:28-33. [ Links ]
54. León-Velarde F. The role of menopause in the development of chronic mountain sickness. Am J Physiol.1997;272:90-94. [ Links ]
55. Arnaud J, Gutiérrez N, Tellez W, Vergnes H. Hematology and erythrocyte metabolism in man at high altitude: an Aymara -Quecchua comparision. Am J Phys Anthropol 1985;67:279-288. [ Links ]
56. Aste-Salazar H, Krundieck CL. Diferenciación de hemoglobinas humanas en las grandes alturas. I. Hemoglobina fetal en recién nacidos y en adultos. Ginecol & Obstet 1971;17:79-102. [ Links ]
57. Chetty KG: Exercise performance of polycythemic chronic obstructive pulmonary disease patients. Chest 1990;98:1073. [ Links ]
58. Queiroz LP, Rapoport AM. High-altitude headache. Curr Pain Headache Rep. 2007;11:293-296. [ Links ]
59. Abdelrahman M. Post-transplant erythrocytosis. Saudi J Kidney Dis Transpl. 2004;15:433-439. [ Links ]
60. Finazzi G. Idiopathic erythrocytosis and other non-clonal polycythemias. Best Pract Res Clin Haematol. 2006;19:471-482. [ Links ]
61. Junpei S. Hemostasis and coagulation at a hematocrit level of 0.85. Blood, 2003;101: 4416-4422. [ Links ]
62. Tekin M. Development of acute coronary syndrome with thrombocythemia. Turk Kardiyol Dern Ars. 2008;36:35-38. [ Links ]
63. Bai J. The risk factors for thrombosis, myelofibrosis and leukemia in patients with PV. Zhonghua. 2007;28:685-688. [ Links ]
64. De Stefano V. Recurrent thrombosis in patients with PV. Haematologica. 2008;93:372-380. [ Links ]
65. Shpilberg O. Polycythemia vera. Gruppo Italiano Studio Policitemia. Ann Intern Med.1995;123:656-664. [ Links ]
66. Johansson PL. An elevated venous haemoglobin cannot be used as a marker. Br J Haematol. 2005;129:701-705. [ Links ]
67. Mossuz P. Diagnostic value of serum Epo level in patients with erythrocytosis. Haematologica, 2004;89:1194-1198. [ Links ]
68. Andrew I. Molecular basis of the diagnosis and treatment of polycythemia vera. Blood, 2006;107: 4214-4222. [ Links ]
69. Ranjan A, Penninga E, Jelsig AM, Hasselbalch HC, Bjerrum OW. Inheritance of the chronic myeloproliferative neoplasms A systematic review. Clin Genet 2012;10.1111/cge.12044. [ Links ]
70. Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012;87:285-293. [ Links ]
71. Gordeuk VR. Congenital polycythemias/erythrocytoses. Haematologica. 2005;90:109-116. [ Links ]
72. Gertz MA, Kyle RA. Hyperviscosity syndrome. J Intensive Care Med. 1995;10:128-141. [ Links ]
73. Tura S. Polycythemic hyperviscosity síndromes. Ric Clin Lab. 1983;13:105-114. [ Links ]
74. Baxter EJ. Acquired mutation of the JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054-1061. [ Links ]
75. Levine RL. Role of JAK2 in the pathogenesis of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673-683. [ Links ]
76. Eid J. Intracellular growth factors in polycythemia vera. Proc Natl Acad Sci USA. 1987;84:532-536. [ Links ]
77. Geissler K. IL-10 inhibits erythropoietin-independent growth of erythroid bursts. Blood. 1998;92:1967-1972. [ Links ]
78. Wolf JT. The effects of IL-1 and IL-4 on the Epo-independent erythroid progenitor in PV. Br J Haematol 1994;88:242-246. [ Links ]
79. Fisher M. Anti-EpoR distinguish EPO-dependent/EPO-independent erythroid progenitors in PV. Blood 1994;84:1982-1991. [ Links ]
80. Rhodes MM. Bcl-xL prevents apoptosis of late-stage erythroblasts. Blood. 2005;106:1857-1863. [ Links ]
81. Dolznig H. Apoptosis protection by the Epo target Bcl-X(L). Curr Biol. 2002;12:1076-1085. [ Links ]
82. Silva M. Erythropoietin can induce the expression of bcl-x(L) through Stat5. J Biol Chem. 1999;274:2165-2169. [ Links ]
83. Simonson T, Prchal J, Ge R. Genetic Evidence for High-Altitude Adaptation in Tibet. Science 2010/ Page 2 / 10.1126/science.1189406. [ Links ]
84. Scortegagna M. The HIF family member EPAS1/HIF-2a is required for normal hematopoiesis in mice. Blood 2003; 1634-1640. [ Links ]
85. Yi X, Liang Y. Sequencing of 50 Human Exomes Reveals Adaptation to High Altitude. Science 2010; 329:75-78. [ Links ]
86. Sokol L. Primary familial polycythemia. Blood. 1995;86:15-22. [ Links ]
87. Arcasoy MO. Erythropoietin hypersensitivity in primary familial polycythemia. Biochim Biophys Acta. 2005;1740:17-28. [ Links ]
88. Forget BG. Familial polycythemia due to truncations of the Epo receptor. Trans Am Clin Climatol Assoc. 2000;111:38-44. [ Links ]
89. Montagna C. In vitro sensitivity of human erythroid progenitors to hemapoietic growth factors. Haematol. 1994; 79:311-318. [ Links ]
90. Dudley JM, Westwood N, Woodcock S, Eridani S. Sensitivity of erythroid progenitors to recombinant growth factors in the diagnosis of myeloproliferative disorders. Int J Cell Cloning 1990;8:199-202. [ Links ]
91. Schwarcz TH , LA Hogan , ED Endean , Roitman IT, Kazmers A , Hyde GL. Las complicaciones tromboembólicas de la policitemia vera: policitemia contra la policitemia fumadores. J Vasc Surg 1993;17:518-522. [ Links ]
92. Varma S, A Sharma, Malhotra P, S Kumari, Jain S, N Varma. Las complicaciones trombóticas de la policitemia vera. Hematología 2008; 13:319-323.
93. Park YH, Huh YE, Kim JS. Oculomotor nerve palsy as an initial manifestation of polycythemia vera. J Clin Neurosci 2012;19:328-330. [ Links ]
94. Larghero J. Farnesyltransferase inhibitor tipifarnib preferentially inhibits in vitro autonomous erythropoiesis of polycythemia vera patient cells. Blood. 2005;105:3743-3745. [ Links ]
95. Matthew SKatz. Potential of statins for cancer chemoprevention and therapy. Nature Clinical Practice Oncology 2005;2:82-89. [ Links ]
96. Mason JC., Statins and their role in vascular protection. Clin Science. 2003;105:251-266. [ Links ]
97. Hamelin BA. Hydrophilicity/lipophilicity: relevance for the pharmacology and clinical effects of HMG-reductase inhibitors. Sci 1998;19:26-37. [ Links ]
98. Ishihara K. Disparity between angiographic regression and clinical event rates with hydrophobic statins. Lancet. 2002;23:729-736. [ Links ]
99. Wolfrum S, Liao JK. Endothelium dependent effects of statins. Arterioscler Throm Vasc Biol. 2003; 23:729-736. [ Links ]
100. Wong WW. HMG-CoA reductase inhibitors and the malignant cell. Leukemia. 2002;16:508-519. [ Links ]
101. Chow SC. Immunomodulation by statins. Arch Immunol Ther Exp (Warsz). 2009 Jul 4. [ Links ]
102. Mason JC., Statins and their role in vascular protection. Clin Science. 2003;105:251-266. [ Links ]
103. Takemoto M. Pleiotropic effects of HMG Co A reductase inhibitors. Arterioscler Thromb Vasc. 2001;21:1712-1719. [ Links ]
104. Sassano A. Regulation ofleukemic cell differentiation by statins. Mol Cancer Ther. 2009; 8:615-625. [ Links ]
105. Burke LP. Statins induce lethal effects in acute myeloblastic leukemia cells. Leuk Lymphoma 2008; 49:832 [ Links ]

