











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkCuadernos Hospital de Clínicas
versión impresa ISSN 1562-6776
Cuad. - Hosp. Clín. vol.62 no.1 La Paz jun. 2021
CASOS CLÍNICOS
Enfermedad de Addison e insuficiencia adrenal aguda: Presentación de un caso y revisión de la literatura
Addison's disease and acute adrenal insufficiency: Case report and literature review
Reyes-Justiniano Andrés1, Beltrán-Luna Eimi C2, Caballero-Chacón Massiel A3
1. Esp. en Medicina Interna, Docente Titular en Farmacología, Facultad de Medicina, UMSA.
2. Estudiante de 4to año de la Facultad de Medicina, UMSA.
3. Estudiante de 4to año de la Facultad de Medicina, UMSA.
Autor para correspondencia: Dr. Andrés Reyes Justiniano, Facultad de Medicina, Enfermería, Nutrición y Tecnología Médica, Universidad Mayor de San Andrés, Av. Saavedra N° 2246 Miraflores, La Paz - Bolivia e-mail: andresreyesfarmacoumsa@gmail.com
RESUMEN
La enfermedad de Addison es una patología endocrinológica ocasionada por la disminución en la secreción de hormonas esteroideas por parte de la corteza adrenal; debida a múltiples etiologías (más comúnmente la tuberculosis en países en vías de desarrollo); con una evolución lenta, insidiosa y progresiva. Pudiendo llegar a una insuficiencia adrenal aguda, misma que puede llegar a ser de extrema gravedad; y en caso de no ser diagnosticada y tratada adecuadamente puede llevar a la muerte. El tratamiento primordial lo constituye la terapia hormonal sustitutiva con fármacos corticoesteroideos.
PALABRAS CLAVE: Insuficiencia adrenal - Ortostatismo - Hiperpigmentación cutánea -Test de cortisol plasmático.
ABSTRACT
Addison's disease is an endocrinological pathology caused by the decrease in the secretion ofsteroid hormones by the adrenal cortex, due to múltiple etiologies (most commonly tuberculosis in deveioping countries); with a slow, insidious and progressive evolution. Being able to reach an acute adrenal insufficiency, which can become extremely serious, and if it is not diagnosed and treated properly, it can lead to death. The primary treatment is hormone replacement therapy with corticosteroid drugs.
KEY WORDS: Adrenal insufficiency - Orthostatism - Skin hyperpigmentation - Plasma cortisol test.
INTRODUCCIÓN
El año 1855, el médico Thomas Addison en Inglaterra, describe por primera vez la signosintomatología de la insuficiencia suprarrenal (actualmente más conocida como insuficiencia adrenal)12.
La enfermedad de Addison (EA) se caracteriza por la insuficiente producción de hormonas corticosuprarrenales de forma crónica, imposibilitando el funcionamiento normal del organismo; esta insuficiencia es ocasionada por la destrucción bilateral de la corteza adrenal, presentando diversas etiologías.34
La insuficiencia adrenal (IA) aguda es un conjunto de síntomas y signos que indican una IA severa, causada por niveles insuficientes de cortisol; desencadenada por situaciones de estrés (cirugías, infecciones o desequilibrio hidroelectrolítico, entre los más destacados).
Puede presentarse como primera manifestación de la EA, no diagnosticada o no tratada, así como descompensación en pacientes sometidos a tratamiento corticoide crónico, tras su supresión brusca. La IA puede ser clasificada en primaria o secundaria según donde se localice el defecto, y ambas formas pueden presentarse de manera aguda o crónica3'56.
Las etiologías más frecuentes de la IA primaria son: adrenalitis autoinmune, adrenalitis tuberculosa, otras infecciones (meningococo, histoplasmosis, micosis y VIH-SIDA), hemorragias, neoplasias (primarias o secundarias), enfermedades por depósito, yatrógenas, congénitas y otras; y dentro de la IA secundaria tenemos como etiologías: la supresión farmacológica tras altas dosis de corticoides y otras como infarto o hemorragia hipofisaria, granulomas e hipofisectomía3'46.
La EA tiene una prevalencia variable según la región, con una edad media de debut entre los 30-80 años. La causa más frecuente de IA primaria en países industrializados es la adrenalitis autoinmune, con predominio del sexo masculino, seguida por el síndrome poliglandular autoinmune (SPA) con una mayor incidencia en el sexo femenino. En países no industrializados la tuberculosis permanece como la principal causa de IA primaria46.
El cuadro clínico es inespecífico y de difícil diagnóstico si no se lo sospecha. La corteza adrenal, al poseer una reserva funcional de hormonas, no manifiesta sintomatología alguna, hasta que existe una pérdida del 80 - 90 % o más de tejido adrenal37.
Entre las principales características clínicas de la IA se mencionan que los síntomas de debilidad, astenia y anorexia se presentan en la práctica totalidad de los casos, siendo frecuentes las náuseas, vómitos y dolor abdominal; el hallazgo físico más importante es la pérdidade peso, siendo el signo común a todos los pacientes, seguida (en un muy alto porcentaje) por la hiperpigmentación y la hipotensión ortostática. Laboratorialmente los trastornos hidroelectrolíticos son de presentación elevada, destacando la asociación de hiponatremia con hiperkalemia y en menor medida la azoemia, también se pueden evidenciar anemia normocítica normocrómica, neutropenia, eosinofilia y linfocitosis relativa6'7.
El diagnóstico se realiza mediante la determinación de cortisol sérico y la hormona adrenocorticotrópica (ACTH) plasmática entre las 6:00 y 9:00 am, las concentraciones normales de cortisol oscilan entre 5 y 20 ug/dL. Un cortisol menor de 3 ug/dL indica la presencia de IA. La determinación de ACTH permite la distinción entre una IA primaria o secundaria68.
El tratamiento de la IA consiste en terapia sustitutiva hormonal, el fármaco de elección es la hidrocortisona, y en casos de IA aguda se administra el mismo fármaco vía parenteral (siendo la vía endovenosa la más utilizada) y reposición hidroelectrolíticaadecuada46.
CASO CLÍNICO
Paciente de sexo femenino, de 39 años de edad, nacida en Oruro y criada en Huanuni, secretaria y estudiante de la Carrera de Trabajo Social en la UMSA. Con el antecedente familiar de padre tratado por tuberculosis pulmonar. La paciente fue diagnosticada de hipotiroidismo a los 28 años de edad, secundario a tiroiditis de Hashimoto; controlada con el tratamiento correspondiente, levotiroxina de 100 microgramos (ug) al día en ayunas; ingresa de emergencia en horario vespertino por aparente síncope, presenciado por ambos padres, con cuadro previo de varios meses de evolución (aproximadamente 9 meses), caracterizados por astenia progresiva, actualmente muy marcada, hiporexia evidente, que conlleva a pérdida de peso de 8 Kg aproximadamente, cefalea biparietal con intensidad EVA (escala visual análoga) de 7 a 8/10; náuseas, que llegan en diversas ocasiones al vómito, dolor abdominal de manera difusa, en ocasiones tipo cólico, en otras no definible, de intensidad variable, despeños diarreicos ocasionales, apetencia selectiva por consumo de alimentos salados desde hace 3 meses y consumo de sal diaria pura durante las últimas 3 semanas; disuria terminal, polaquiuria y estranguria desde hace 14 días aproximadamente y finalmente, dolor lumbar izquierdo con intensidad EVA 8/10, cefalea holocraneana y sensación febril franca, no cuantificada las ultimas 48 horas.
Al examen físico se evidencia: Hiperpigmentación en rostro, dentro de cavidad bucal en región gingival, cuello, regiones axilares, genitales externos, codos, rodillas, tobillos y articulaciones interfalángicas de ambas manos. Presencia de puntos ureterales anteriores alto y medio izquierdos presentes, puñopercusión lumbar homolateral positiva, y punto costovertebral izquierdo positivo. Reporta los siguientes signos vitales: Tensión arterial (TA): 98/54 mm de Hg en brazo derecho y 100/56 mm de Hg en brazo izquierdo, ambos en decúbito dorsal, la TA disminuye al ortostatismo, FC: 104 lat/min, FR; 24 resp/min, temperatura oral: 37,9 C°. Peso: 58,3 Kg (pesaba 66 kg hace 8 meses), Talla: 1,64 m. IMC: 21,67 Kg/m2.
Los laboratorios realizados al ingreso evidenciaron: Hemograma: Anemia leve normocítica normocrómica (Hb de 12,7 g/dL y Hto 48 %), leucocitosis con neutrofilia (GB: 14,200, Neutrófilos: 82 %), EGO: Con datos de infección de vía urinaria (Nitritos positivos, GB: 58 a 60 x campo, bacterias abundantes y presencia de piocitos). Na: 128 mEq/L y K: 5,4 mEq/L, glucemia al azar: 74 mg/dL, creatinina: 1,5 mg/dL y urea: 69 mg/dL.
Se diagnostica pielonefritis aguda y se inicia tratamiento con antibióticos empíricos (cefotaxima vía parenteral), además de reposición volumétrica con hidratación y expansores sin elevación de la tensión arterial. Ante la sospecha clínico laboratorial de IA aguda, se realizan los siguientes laboratorios en ayunas: TSH: 1,8 (N: 0,3 a 4,0), T3: 106 (N: 81 a 178) Y T4: 7,40 (N: 4,5 a 12,5), evidenciando hipotiroidismo controlado, Cortisol plasmático a las 8:00 am con un resultado de: 2,6 ug/dL (N: 3-20 ug/dL), con lo que se corroboró IA aguda.
Se instaura tratamiento hormonal sustitutivo en base a hidrocortisona parenteral a dosis diarias decrecientes, con restitución de signos vitales y parámetros laboratoriales (incluidos los hidroelectrolíticos) en 48 horas.
Posteriormente, durante su internación se corrobora por medios imagenológicos la calcificación y atrofia de ambas glándulas adrenales sugiriendo una probable etiología tuberculosa. No se realizó dosificación de ACTH, como tampoco anticuerpos contra corteza adrenal.
REVISIÓN DE LA LITERATURA
INSUFICIENCIA ADRENAL Y ENFERMEDAD DE ADDISON
DEFINICIÓN
La IA es una patología ocasionada por la disminución en la secreción de hormonas esteroideas por parte de la corteza adrenal, caracterizada por el déficit predominante de glucocorticoides. Esta puede ser clasificada en primaria, siendo la causa más frecuente la adrenalitis autoinmune (y la tuberculosis en países en vías de desarrollo); o secundaria, por alteración en la secreción de la ACTH o de la hormona liberadora de ACTH (CRH), cuya causa más común es la suspensión brusca de glucocorticoides. Al ser una patología infradiagnosticada, los pacientes presentan deterioro en la calidad de vida y el riesgo de presentar una IA aguda que podría llegar a ser letal.69
ETIOLOGÍA
La causa más frecuente de IA primaria en los países industrializados, es la adrenalitis autoinmune, que constituye el 80 - 90% de todos los casos. En un 40% puede presentarse de forma aislada con predominio del sexo masculino y en el 60% restante formar parte de un SPA con una mayor incidencia en el sexo femenino. En países en vías de desarrollo (como en Bolivia) la tuberculosis es aún la principal causa de IA primaria, la adrenalitis tuberculosa suele provenir de un foco tuberculoso extrapulmonar, por lo general genitourinario. Otras causas infecciosas son las micóticas y virales (particularmente en pacientes con VIH-SIDA).
La más destacada dentro de las congénitas es la adrenoleucodistrofia; también se presentan por origen quirúrgico, tumoral o secundario a uso de fármacos inductores (ej. Rifampicina); finalmente se pueden producir hemorragias (ej. Síndrome de Waterhouse-Friderichsen) o trombosis de las glándulas adrenales por diátesis hemorrágica, uso de anticoagulantes orales, estados de hipercoagulabilidad o, más raramente, síndrome antifosfolipídico2'3'6.
En la IA secundaria la causa más común es la suspensión brusca del tratamiento con glucocorticoides, posterior a su uso por un período prolongado. La administración exógena y prolongada de glucocorticoides induce atrofia de las células corticotropas y supresión del eje hipotálamo-hipófisis-adrenal (HHA). Dentro de los SPA, el SPA tipo I (o síndrome de Whitaker) es poco común, se presenta más en la infancia, cursa principalmente con hipoparatiroidismo, enfermedad de Addison, candidiasis en piel y mucosas. La IA se desarrolla en el 80% de los pacientes con SPA tipo I y el 18% presenta diabetes tipo 1. El SPA tipo II (o síndrome de Schmidt), de presentación más común en mujeres de edad media, más frecuente que el anterior, corresponde a la asociación de Addison con hipotiroidismo autoinmune y/o diabetes tipo 1. Pudiendo asociarse: falla gonadal, vitíligo, anemia perniciosa, enfermedad celíaca o hepatitis autoinmune. Cabe acotar que un paciente que debuta con EA tiene hasta un 50% de posibilidad de tener un SPA. Siguen en frecuencia etiológica, la presencia de tumores de la región hipotálamo-hipofisiaria, así como las opciones terapéuticas disponibles para los mismos (cirugía y radiación) como generadores de IA secundaria, finalmente, hasta un tercio de las IA no llegan a tener un diagnóstico etiológico (ver cuadral f-6-7-10.
Cuadro N° 1. Causas más frecuentes de Insuficiencia Adrenal
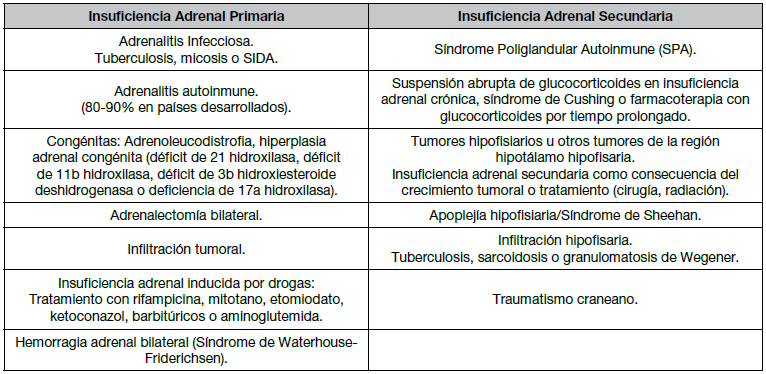
Modificado de Aguirre Miguel A, Luna Magda, Reyes Yubríangel, Gómez-Pérez Roald, Benítez Isabel. Diagnóstico y Manejo de la Insuficiencia Adrenal. Rev. Venez. Endocrinol. Metab. [Internet]. 2013 oct. [citado 2021 Ene 21]; 11 (3): 157-167.
MANIFESTACIONES CLÍNICAS
Los síntomas más frecuentes de la IA crónica son astenia marcada predominantemente vespertina, fatiga, debilidad, pérdida de peso, disnea, y manifestaciones gastrointestinales como hiporexia, lo que ocasiona una pérdida de peso que va desde los 2 hasta los 15 kg. Además de avidez por la sal y tendencia a la hipodipsia (ingestión escasa de líquidos), náuseas, vómitos, y dolores abdominales, que en las crisis addisonianas pueden incluso emular al abdomen agudo. Puede haber constipación e hipomotilidad intestinal y en menor medida diarrea, en presencia de la misma, es necesario descartar enfermedad celíaca2'4'12-13.
También aparecen síncope, taquicardia, palpitaciones y vértigo. En reposo el pulso es normal, pero los mínimos esfuerzos o emociones desencadenan taquicardias desproporcionadas.4
El hallazgo físico más importante es la pérdida de peso, siendo el signo común a todos los pacientes, seguido en un muy alto porcentaje por la hiperpigmentación y la hipotensión arterial. La hiperpigmentación es más acentuada en áreas expuestas al sol y a la presión (pliegues palmares, codos, rodillas, dedos del pie, axilas, areolas, mucosa de cavidad oral y genitales externos). La hipotensión arterial es típicamente ortostática y se presenta vitíligo en menor medida2'14.
La evolución suele ser lenta, insidiosay progresiva, el proceso destructivo puede prolongarse por años, incluso décadas (ver cuadro N° 2)11.
Cuadro N° 2. Características clínicas y laboratoriales de la insuficiencia adrenal
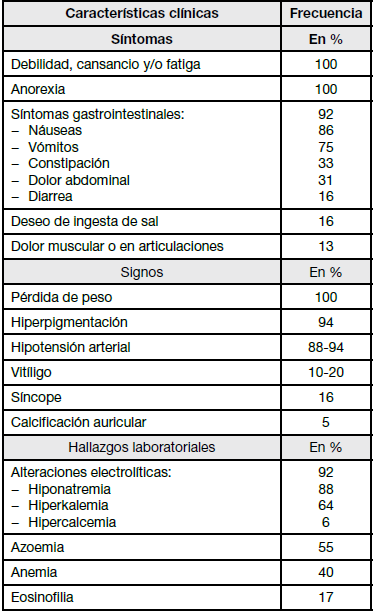
Modificado de Aguirre Miguel A, Luna Magda, Reyes Yubríangel, Gómez-Pérez Roald, Benítez Isabel. Diagnóstico y Manejo de la Insuficiencia Adrenal. Rev. Venez. Endocrinol. Metab. [Internet]. 2013 Oct [citado 2021 Ene 21]; 11(3): 157-167.
ESTUDIOS DE LABORATORIO
Puede observarse que el déficit mineralocorticoide (hipoaldosteronismo) induce una pérdida salina urinaria con retención de potasio. La hiponatremia potenciada por la incapacidad para excretar agua libre, junto con la hiperpotasemia mencionada, además de retención nitrogenada y acidosis metabólica contribuyen a los trastornos de la motilidad de la musculatura lisa y estriada, dando lugar a las manifestaciones gastrointestinales. La hipercalcemia es otro signo acompañante que puede alcanzar valores similares a la crisis hipercalcémica. El déficit de cortisol, como hormona contrarreguladora, cursa con frecuencia con hipoglucemia.(12)
La acidosis metabólica es hiperclorémica; aumenta la urea por depleción de volumen; existe anemia que puede ser normocítica normocrómica, o macrocítica por anemia perniciosa asociada; neutropenia, eosinofilia y linfocitosis. (ver cuadro 2)2, 4
DIAGNÓSTICO
La medición del cortisol sérico se recomienda realizarla entre las 6:00 y las 9:00 de la mañana debido al carácter pulsátil de la hormona, dándose los niveles más altos en las primeras horas del día. Las concentraciones normales de cortisol oscilan entre 5 y 20 ug/dL. Un cortisol menor de 3 ug/ dL indica la presencia de IA y valores superiores a los 18 ug/dL excluyen el diagnóstico. En caso de duda, realizar el test de estimulación con ACTH intravenosa, el mismo permite la distinción entre una IA primaria o secundaria, siendo primaria si existen concentraciones superiores a los 100 pg/mL de ACTH, a diferencia de ACTH baja o inapropiadamente normal en relación a las concentraciones de cortisol, en casos de IA secundaria876.
La positividad de anticuerpos anti adrenales (también conocidos como anticuerpos contra corteza adrenal), asociados a una tomografía axial computarizada (TAC) abdominal, que evidencie glándulas normales o atróficas, caracterizan a la causa autoinmune; también es importante la asociación con otras enfermedades autoinmunes2.
Si se sospecha de procesos infecciosos, infiltrativos o neoplásicos debe ser realizada también una TAC abdominal, donde típicamente en la tuberculosis, se observan calcificaciones, sobre glándulas inicialmente aumentadas de tamaño que luego se atrofian. Por su parte, ante la sospecha de IA secundaria de origen desconocido, la realización de una Resonancia Magnética Nuclear (RMN) de la región hipotálamo hipofisaria es el método ideal para la identificación de lesiones ocupantes de espacio6.
TRATAMIENTO
En condiciones fisiológicas, la secreción de cortisol presenta un ritmo circadiano alcanzando un pico entre las 6:00 y 9:00 am y un nadir entre las 11:00 pm y 2:00 am, con una tasa de producción diaria de cortisol de 5 a 10 mg/m2 de superficie corporal que puede incrementarse ante situaciones de estrés, por lo tanto, la terapia sustitutiva debe tratar de simular este patrón fisiológico. Los objetivos de la misma incluyen alcanzar una expectativa con calidad de vida adecuada, evitando los efectos adversos de la terapia de reemplazo hormonal.615
El tratamiento se realizará en pacientes sintomáticos, no en aquéllos en los que tan sólo se aprecian anormalidades hormonales. Se desea reemplazar el glucocorticoide faltante y el mineralocorticoide, en caso necesario. El fármaco de elección para el tratamiento sustitutivo del déficit hormonal es la hidrocortisona, que presenta un efecto dual, tanto glucocorticoide como mineral o corticoide. La dosis de mantenimiento es de 20-25 mg/día de hidrocortisona y 25-35 mg/día de cortisona, no olvidando la forma de administración fraccionada: 2/3 por la mañana y 1/3 por la tarde, emulando el ciclo circadiano.4'6'7'16
La alternativa terapéutica similar en resultados, es la asociación de dexametasona (2,5-7,5 mg/día) o de prednisona (0,25-0,75 mg/día) con un mineralocorticoide, por ejemplo, la fludrocortisona.6
En pacientes con IA primaria, cuando a pesar de dosis óptimas de hidrocortisona persisten síntomas como hipotensión u ortostatismo, se indicará tratamiento con mineralocorticoides ya que el Sistema Renina Angiotensina Aldosterona también se encuentra afectado. Se emplea la fludrocortisona a dosis de 0,05 a 0,2 mg/día en una sola toma6.
En pacientes cuya hiperpigmentación no evidencia mejoría a pesar de dosis óptimas de hidrocortisona, la administración de glucocorticoides de acción prolongada puede ofrecer una mejor cobertura durante la noche con corrección del cuadro6.
En los cuadros clínicos en los que no se pueda garantizar la absorción del 100% del fármaco (como en diarreas) la dosis podría administrarse por vía intramuscular 50 mg cada 12 horas hasta su total recuperación (ver cuadro 3).46
Cuadro N° 3. Comparación de potencias, dosis equivalente y vida media de los principales corticoides
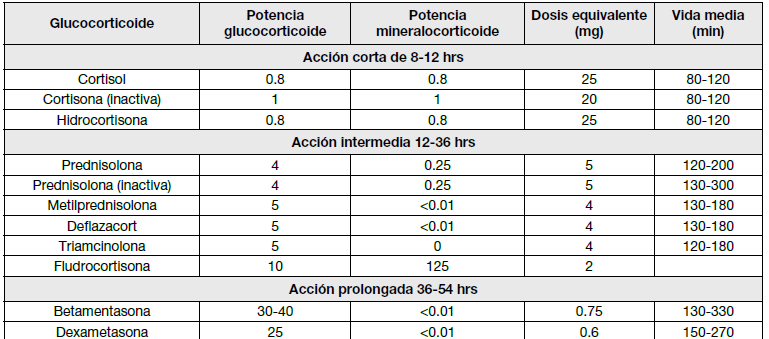
Modificado de Vera Carrasco O, Botelho Perpich A, Segales Pabón F, Vera Navarro L, Reyes Justiniano A, Peñafiel Rodríguez L et al. Texto de la Cátedra de Farmacología. Primera edición La Paz - Bolivia: Dr. Vera Carrasco; 2020; Sección VI. Farmacología Endocrinología y del Metabolismo. Cap. 2: Corticosteroides, p. 442-449.
INSUFICIENCIA ADRENAL AGUDA
La IA aguda, es un cuadro clínico abrupto que compromete la vida del paciente por lo que es considerado una emergencia endocrinológica. Ya sea frente a una hemorragia, necrosis o trombosis adrenal, o cuando a un paciente portador de enfermedad crónica se agrega una situación de estrés desencadenante, el cuadro evoluciona a ser de extrema gravedad, que de no ser diagnosticado adecuadamente y consecuentemente tratado puede llevar a la muerte26.
ETIOLOGÍA
La IA aguda es un conjunto de síntomas que indican una IA severa causada por niveles insuficientes de cortisol. Puede presentarse como primera manifestación de la IA crónica (EA), no diagnosticada o no tratada (IA primaria), o como descompensación en pacientes sometidos a tratamiento corticoideo crónico, tras la supresión brusca de la misma (siendo esta la más frecuente de sus presentaciones), disminución de la dosis de glucocorticoides o ausencia de ajuste en la dosis ante situaciones de estrés como pancreatitis aguda grave, infecciones severas, quemaduras extensas o cirugías mayores3.
La falta de terapia de reemplazo posterior a la corrección de un síndrome de Cushing puede desencadenar una IA aguda debido a la supresión del eje HHA por las altas concentraciones de cortisol durante un largo período. La hemorragia adrenal secundaria a procesos infecciosos o coagulopatías, así como necrosis hipofisaria debida a hemorragia postparto (síndrome de Sheehan), constituyen causas de IA aguda en pacientes sin deficiencia de glucocorticoides previamente conocida6.
La administración de determinados fármacos puede desencadenar una crisis adrenal, como el ketoconazol, la rifampicina o la fenitoína (entre otros), que actúan alterando el metabolismo de los corticoides y conducen a una insuficiencia adrenocortical en pacientes con dosis de sustitución fijas3.
En un estudio con 444 pacientes, el 42% informó al menos una crisis desde el inicio de su patología; la frecuencia de las crisis se calculó en 6,3/100 pacientes año. Los pacientes con IA primaria tenían una probabilidad ligeramente mayor de sufrir una crisis que aquellos con IA secundaria. Los desencadenantes más frecuentes de la crisis adrenal son las causas infecciosas.17
CLÍNICA Y DIAGNÓSTICO
Debe sospecharse en todo paciente en estado crítico, con hipotensión arterial refractaria a la expansión con fluidos y drogas presoras (característicamente la tensión arterial está por debajo de 110/70 mm Hg), si existiese hipertensión arterial se debe replantear el diagnóstico de IA, la clínica presenta alteraciones como astenia y anorexia marcadas, pérdida de peso importante, hiperpigmentación, vitíligo, dolor abdominal, náuseas y vómitos, estos últimos pueden hacer pensar en abdomen agudo quirúrgico, al cuadro clínico se añaden manifestaciones laboratoriales como anemia, hiponatremia, hiperkalemia y azoemia entre las más destacadas, siendo menos frecuentes la eosinofilia, linfocitosis, hipoglicemia e hipercalcemia. El síncope puede ocurrir en el 16% de los casos23.
Las alteraciones electrocardiográficas pueden mostrar ondas T picudas por hiperkalemia o un intervalo QT corto por hipercalcemia. Se debe solicitar una radiografía de tórax, análisis de orina y hemocultivos para evaluar cualquier infección17.
Una TAC de abdomen puede mostrar hemorragia en las glándulas adrenales, calcificación en las mismas (tuberculosis) o metástasis. En casos de IA secundaria, una TAC de encéfalo, puede mostrar la destrucción de la hipófisis (es decir, el síndrome de la silla turca vacía) o una lesión de masa hipofisaria (ver cuadro 2)17.
MANEJO
El tratamiento de la IA aguda, como el de cualquier emergencia médica, debe iniciarse ante su sola sospecha, pues la demora hasta la confirmación diagnóstica mediante la determinación hormonal aumenta notablemente la morbimortalidad del paciente. Se basa en la adopción de una serie de medidas generales y en la instauración de la terapia hormonal sustitutiva.
El tratamiento de la depleción hidrosalina y la hiponatremia se realiza mediante administración de solución fisiológica, alternando con solución glucosada al 5%, a razón de 1.000 ml/h, durante las primeras 4 h. Después, se perfunde el mismo tipo de líquido a un ritmo de 1.000 ml/6 h.
La hipoglucemia se corrige, generalmente, con la administración de solución glucosada al 5% utilizado para la reposición hídrica.
TRATAMIENTO HORMONAL SUSTITUTIVO
A continuación, utilizamos la terapéutica descrita en el libro de Medicina de Urgencias y Emergencias (Jiménez Murillo y Montero Pérez), por ser la mejor descrita y completa. Se basa en la administración de glucocorticoides por vía intravenosa. Pudiendo utilizar una de las dos pautas terapéuticas a continuación, teniendo en cuenta que, si el paciente ya está diagnosticado y hay hiperpotasemia, es preferible la primera, por su mayor efecto mineralocorticoide; por el contrario, si el enfermo no está diagnosticado de IA, es mejor administrar dexametasona, ya que esta no interfiere con las pruebas diagnósticas hormonales a ser realizadas posteriormente.
La hidrocortisona se administra de la siguiente forma: Una dosis inicial de 100 mg, en bolo intravenoso, seguida, las primeras 24 h, de 100 mg/6 h por vía intravenosa, diluidos en la solución elegida para la corrección de la depleción hidrosalina, o en perfusión continua, para lo que se diluyen 400 mg (4 ampollas de 100 mg) en 500 mi de suero fisiológico, y se perfunde «en Y» con la reposición de volumen, a un ritmo de 7 gotas/ min (21 ml/h).
Durante el segundo día se administran 100 mg/8 h por vía intravenosa, y se reduce progresivamente la dosis en los días sucesivos (cada 24 h) a 100 mg/12 h y a 50 mg/12 h durante el tercer y cuarto días, respectivamente; en general, la hidrocortisona por vía oral puede empezar a administrarse al quinto día de haber iniciado el tratamiento intravenoso.
La segunda pauta terapéutica consiste en utilizar dexametasona en dosis inicial de 4-8 mg, en bolo intravenoso, seguida de 4 mg/4-6 h por la misma vía.
Junto a la hidrocortisona o la dexametasona, se debe administrar mineralocorticoides, como fludrocortisona, en una dosis matutina de 0,05 mg, que puede incrementarse hasta alcanzar un máximo de 0,2 mg cada 24 h por vía oral. Si se ha elegido la hidrocortisona como tratamiento inicial, la fludrocortisona se administra cuando la dosis diaria de hidrocortisona sea inferior a 100 mg, generalmente desde el comienzo de la administración por vía oral. Si por el contrario se ha elegido la dexametasona, la administración de fludrocortisona debe hacerse desde el primer día.18
Finalmente, para evitar una IA aguda, en pacientes bajo tratamiento corticoideo prolongado podemos seguir las siguientes recomendaciones: Nunca suspender los glucocorticoides de forma brusca, calcular dosis Kg/peso y utilizar la menor dosis Kg/peso efectivo.(19)
CONCLUSIÓN
La enfermedad de Addison y la insuficiencia adrenal aguda, como complicación de la primera (debido a múltiples causas), es considerada como una patología secundaria a destrucción de las glándulas adrenales, de etiología tuberculosa en Bolivia, siendo la etiología autoinmune la más común en países desarrollados. El conocimiento profundo de la clínica, laboratorio, estudios complementarios y sobre el comportamiento de la insuficiencia adrenal, son las bases para el adecuado diagnóstico. El tratamiento precoz (sin confirmación de cortisol plasmático) con reposición hidroelectrolítica adecuada y terapia de reemplazo con corticoesteroides, disminuyen la elevada mortalidad. La necesidad de estudios sobre origen etiológico en nuestro medio es imperativa.
REFERENCIAS
1. Rojas Paz M. Crisis Addisoniana aguda secundaria a suspensión abrupta de corticoides [Licenciatura]. Universidad Técnica de Ambato, Facultad de Ciencias de la salud; 2019. [ Links ]
2. Loto MG, Misiunas A, Curria M, Finn BC, Bruetman JE, Young P. Biografía de Thomas Addison (1793-1860) y revisión breve de la insuficiencia suprarrenal primaria, 2013; VIII (1280). [ Links ]
3. Estaben Boldova V, Llannuzzelli Barroso C. López Mas C, Sanchis Yago B.Crisis addisoniana como diagnóstico de enfermedad de Addison. Revista Atalaya Médica, No 4/2013. p. 55-58 [ Links ]
4. Candel González F. J., Matesanz David M., Candel Monserrate I. Insuficiencia corticosuprarrenal primaria: Enfermedad de Addison. An. Med. Interna (Madrid). 2001; 18(9): p. 48-54. Disponible en: http://scielo.isciii. es/scielo.php?script=sci_arttext&pid=S0212-71992001000900011&lng=es. [ Links ]
5. FaresAdilB, SantosRómuloAdos. Conductprotocolinemergency:Acuteadrenalinsufficiency. Rev. Assoc. Med. Bras. [Internet]. 2016; 62(8): p. 728-734. Disponible en: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0104-42302016000800728&lng=en. http://dx.doi.org/10.1590/1806-9282.62.08.728. [ Links ]
6. Aguirre Miguel A, Luna Magda, Reyes Yubriangel, Gómez-Pérez Roald, Benítez Isabel. Diagnóstico y manejo de la insuficiencia adrenal. Rev. Venez. Endocrino!. Metab. [Internet]. 2013; 11(3): p. 157-167. Disponible en: http://ve.scielo.org/scielo.php?script=sci_arttext&pid=S1690-31102013000300007&lng=es. [ Links ]
7. Wang-Zúñiga P, Chen-Ku Chih Hao. Diagnostico y tratamiento de la enfermedad de Addison; ejemplos de su manejo clínico. Revista médica de la universidad de Costa Rica, 2007:1(1): p. 35-52. Disponible en: https://revistas.ucr.ac.cr/index.php/medica/article/view/7872 [ Links ]
8. Muñoz A., Oñate J., Mané J. M., Rubio I., Fernández R., Barceló J. R. et al. Addisonian crisis as initial manifestation of adrenal insufficiency in a patient with lung cáncer. An. Med. Interna (Madrid). 2001; 18(1): p. 35-37. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0212-71992001000100010&lng=es. [ Links ]
9. Rodríguez Rodríguez M., Columbié Singh A., Delgado Matos M., Wilson Chibas L., Otamendy Fernandez C, Riverón Núñez R., Rodríguez Lobaina X. Enfermedad de Addison. Informe de un caso. Revista información científica [Internet]. 2008;59(3): Recuperado de: https://www.redalyc.org/articulo.oa?id=551757323012 [ Links ]
10. Soria Oviedo P., Morales Calderón M., Alfonso de León J.A., Álvarez Escobar M.C., Torres Álvarez A. Hiperpigmentación generalizada. A propósito de un caso. Rev. Med. Electrón. [Internet]. 2013; 35(3): p. 287-295. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1684-18242013000300009&lng=es. [ Links ]
11. López Bilbao la Vieja, I. Enfermedad de Addison: presentación de dos casos clínicos. Cuad. Hosp. Clin, 1998. p. 72-74. Disponible en: http://biblioteca.fment.umsa.bo/docs/tc/chc1998440111.pdf [ Links ]
12. Rosell Vergara Eduardo. Insuficiencia suprarrenal crisis Addisoniana. Málaga. Disponible en: http://www. medynet.com/usuarios/jraguilar/Manual%20de%20urgencias%20y%20Emergencias/insufren.pdf [ Links ]
13. Narváez-Casillas VE, Vargas-Hernández A. Enfermedad de Addison en un adolescente masculino. Rev. del Hospital Juárez de México 79.2 2012: p. 119-124. Disponible en: https://www.medigraphic.com/cgi-bin/ new/resumenl.cgi?IDARTICUL0=42628 [ Links ]
14. ¡piales Miranda MA. Análisis de caso clínico sobre: "Enfermedad de Addison" [tesis de grado]. Ambato, Ecuador: Universidad técnica de Ambato, Facultad de ciencias de la salud; 2015. [ Links ]
15. Hahner S, Allolio B. Therapeutic managnment of adrenal insufficiency. Best Pract Res Clin Endocrino! Metab 2009; 23: p. 167-179 [ Links ]
16. Prieto de Paula J. M., Aliaga y Montilla M. A., Alonso Fernández J. I., Martín Serradilla J. I., Relea Sarabia A., Villamandos Nicas V. Varón de 69 años con afectación del estado general, hiperpigmentación, lesión cutánea y respuesta deficiente al tratamiento. An. Med. Interna (Madrid). 2007;24(12):p. 599-601. Disponible en: http://scieloisciii.es/scielo.php?script=sci_arttext&pid=S0212-71992007001200009&lng=es. [ Links ]
17. Rathbun Kimberly M., Nguyen Minhthao, Singhal Mayank. Addisonian Crisis. StatPearls - NCBI Bookshelf. 2020. p. 1-9 [ Links ]
18. Jiménez Murillo L, Montero Pérez F. Medicina de urgencias y emergencias. 6th ed. España: Elsevier; 2019; p. 479-480. [ Links ]
19. Vera Carrasco O, Botelho Perpich A, Segales Pabón F, Vera Navarro L, Reyes Justiniano A, Peñafiel Rodríguez L et al. Texto de la Cátedra de Farmacología. Primera edición La Paz - Bolivia: Dr. Vera Carrasco; 2020; Sección VI. Farmacología Endocrinológica y del Metabolismo. Cap. 2: Corticosteroides, p. 442-449. [ Links ]
20. Cálculo de Equivalencias de Corticoides [homepage en la Web]. R.Pitarch Flors, 2011. Disponible en: https://www.rccc.eulppclcalculadoraslcorticoides.htm [ Links ]

