










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO  uBio
uBio Compartir

 Permalink
PermalinkCuadernos Hospital de Clínicas
versión impresa ISSN 1562-6776
Cuad. - Hosp. Clín. vol.60 no.1 La Paz jun. 2019
CASOS CLÍNICOS
Trisomía 9: Reporte de un caso
Trisomy 9: a case report
Luna-Barrón B1, Taboada-López G1, Siacar-Bacarreza S2, Lafuente-Álvarez E3, Rada-Tarifa A4, Darinka Contreras-Castro T5, Burgos-Zuleta J L6
1 Médico Genetista. Instituto de Genética de la Universidad Mayor de San Andrés
Autor para correspondencia: Dra. Beatriz Luna Barrón, Instituto de Genética de la Universidad Mayor de San Andrés, La Paz Bolivia, blunab3@gmail.com
2Pediatra Endocrinóloga. Servicio de Endocrinología Infantil, Hospital Materno Infantil de la CNS
3Bioquímica Genetista. Instituto de Genética de la Universidad Mayor de San Andrés
4Bioquímica Farmacéutica. Instituto de Genética de la Universidad Mayor de San Andrés
5Médico Cirujano. Instituto de Genética de la Universidad Mayor de San Andrés
6Servicio de Imagenología, Caja de la Banca Privada de Salud.
Resumen
La trisomía 9 es una enfermedad rara, que ha sido descrita por primera vez en 1970, a la fecha existen más de 150 casos reportados, caracterizados por dismorfias faciales, anomalías congénitas y retraso en el desarrollo psicomotor y/o discapacidad intelectual. Este es el primer caso reportado en nuestra población en un infante de sexo masculino con peso y talla bajos, fisura labiopalatina y retraso madurativo en varias áreas del desarrollo, en quien el cariotipo mostró un mosaico cromosómico con el 70% de sus células con la trisomía del cromosoma 9. El asesoramiento genético en estos casos es de vital importancia para orientar a los padres sobre posibles causas y explicar sobre la condición genética, su manejo y establecer pautas de seguimiento para hacer prevención terciaria.
INTRODUCCIÓN
La carga genética (ploidia) en la especie humana corresponde a 23 pares de cromosomas, de los cuales 22 son autosomas y 2 son gonosomas. Las cromosomopatías son alteraciones en el número y/o la estructura de los cromosomas, entre las cuales están las aneuploidías, definidas como la presencia o ausencia de uno o más cromosomas (no múltiplo de 23), dentro de este grupo, las trisomías son las más frecuentes, como el síndrome de Down (trisomía 21), el síndrome de Edwards (trisomía 18) y el síndrome de Patau (trisomía 13). De acuerdo con la literatura (1, 2) la cuarta trisomía viable más frecuente corresponde a la presencia de tres cromosomas en el par 9 (trisomía 9 parcial (9p) o completa), antiguamente conocido como síndrome de Retorhé. Este hecho está en relación al concepto de mosaico cromosómico, en el que coexisten 2 líneas celulares con cargas genéticas distintas en un mismo individuo, o casos en los cuales existen aneusomías segmentarias (trisomía parcial de sólo un fragmento del cromosoma 9). El primer caso de trisomía 9 fue descrito en 1970, desde entonces existen más de 150 pacientes reportados en la literatura1. A diferencia de las trisomías más frecuentes (libres) la mayor parte de los casos se debe a translocaciones balanceadas en los padres.
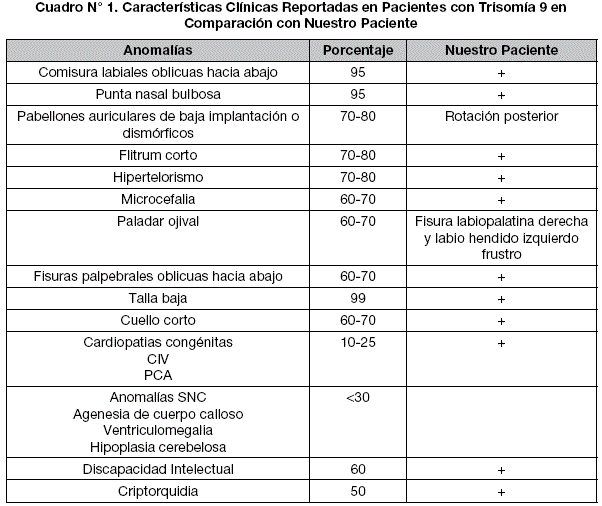
Clínicamente esta cromosomopatía se caracteriza por rasgos faciales dismórficos, malformaciones congénitas y discapacidad intelectual (Cuadro N°1)2. La variabilidad fenotípica se debe a que el tamaño de cromosoma involucrado es diferente en cada caso. La presencia de discapacidad intelectual de grado variable, así como de otras características físicas tales como el hipertelorismo ocular, comisura bucal hacia abajo, nariz bulbosa y la hipoplasia de la falange distal, en presencia de una trisomía del brazo corto, con o sin presencia de parte del brazo largo, han hecho que esta trisomía sea reconocida como Síndrome 9p (como punto crítico)2,3,4. La gran mayoría de los embarazos con trisomía 9 terminan en un aborto espontáneo y se diagnostican sólo si se realiza un cariotipo rutinario de los restos ovulares o de líquido amniótico. Los casos que sobreviven a la etapa postnatal son mosaicos cromosómicos de distintas proporciones5.
Su diagnóstico laboratorial se basa en el estudio de cariotipo con bandeo GTG (con la utilización de tinción Giemsa y tripsina), y se puede complementar con hibridación fluorescente in situ (FISH), o por hibridación genómica comparativa (CGH), que permite determinar la región específica del cromosoma 9 implicada y su tamaño3.
El interés de este caso clínico busca reportar el primer caso en Bolivia, así como orientar el diagnóstico para el asesoramiento genético de estas familias y su seguimiento precoz, pese a su baja frecuencia. Las exploraciones complementarias básicas recomendables para definir la expresión fenotípica en cada caso concreto son: estudio óseo radiológico, ecografía abdominal, pruebas de neuroimagen y valoración del desarrollo psicomotor por un equipo multidisciplinario en el que intervenga el neuropediatra, el fisioterapeuta y los distintos profesionales del equipo de atención temprana5, 6, 7.
CASO CLÍNICO
Masculino de 5 años de edad, producto de 2da gestación, padres no consanguíneos, sin antecedentes de exposición a teratógenos durante su gestación. Nació por cesárea debida a preeclampsia, siendo prematuro (30 semanas de gestación SDG). Sin antecedentes familiares de importancia. Antecedentes patológicos de fisura labiopalatina derecha corregida a los 3 años, criptorquidia corregida, retraso del desarrollo psicomotor, retraso en el desarrollo del lenguaje, talla baja y falla de medro.
A la exploración física talla <p5, peso <5p, perímetro cefálico p50, trigonobraquicefalia, frente alta, facies dismórfica, fisuras palpebrales oblicuas hacia abajo, hipertelorismo, telecanto, cejas escasas. Puente nasal ancho, nariz con punta bulbosa, filtrum corto, con cicatriz de fisura labiopalatina (derecha reparada, izquierda frustra), comisuras labiales oblicuas hacia abajo, maxilar inferior en punta, pabellones auriculares normoimplantados, con rotación posterior, cuello corto, tórax con pectus excavatum inferior, genitales masculinos sin alteraciones, miembros superiores e inferiores con uñas hiperconvexas. Mancha hipocrómica grande de bordes irregulares en hemitórax izquierdo. Marcha conservada, no habla, no se comunica con el entorno.
En el estudio citogenético en sangre periférica del paciente se observó un 30% de metafases con carga cromosómica normal (46, XY) y un 70% de las metafases con cariotipo 47,XY,+9 (Fig. N°1). Correspondiente a un mosaico de la trisomía del cromosoma 9 en un individuo de sexo masculino. Para complementar la evaluación citogenética se realizó la técnica de bandeo CTC que permitió observar la región centromérica (polimórfica) de los cromosomas 9, siendo visualizados en triple dosis en un 70% de las metafases analizadas. No se aplicaron técnicas de citogenética molecular por escases de recursos. En el asesoramiento genético se sugirió cariotipo a ambos padres.

DISCUSIÓN
Este es el primer caso de trisomía 9 reportado en nuestra población, en relación a su baja frecuencia (siendo parte del grupo de enfermedades raras o poco frecuentes)8. La mayor parte de la literatura sobre esta cromosomopatía reporta casos de diagnóstico prenatal, con una supervivencia que no supera los 4 meses de vida (trisomía 9 completa) en el 92% de los casos y el 48% en los mosaicos9, es importante mencionar que la mayor esperanza de vida en los mosaicos, no se relaciona con el porcentaje de células con trisomía ya que está última no predice la mayor o menor severidad del fenotipo10, 11.
Las trisomías se relacionan con errores durante la división del óvulo o el espermatozoide de los progenitores (no disyunción o rezago anafásico en meiosis) o por alteraciones en la segregación cromosómica en las etapas más incipientes de la gestación (mitosis), este último concepto en probable relación al mosaicismo cromosómico observado en el paciente.
La talla baja del paciente y las dismorfias faciales fueron el motivo de consulta y solicitud de cariotipo, lo que concuerda con las indicaciones de realización de este tipo de estudios genéticos dada la amplia gama de diagnósticos diferenciales con los signos descritos. Los puntos clave para sospechar una condición sindrómica son las dismorfias faciales que se presentan con retardo psicomotor o discapacidad intelectual y anomalías congénitas. Estos son los 3 pilares fundamentales que nos dan la sospecha inicial de una cromosomopatía o una genopatía.
En relación a la parte molecular, existen muchos genes localizados en el cromosoma 9, su expresión patológica puede explicar el fenotipo característico de este síndrome, por ejemplo la alteración del gen que codifica para el factor de transcripción tiroidea (FOX2) ubicado en 9q 22.33 se ha relacionado con fisuras labiopalatinas12 (presente en nuestro paciente), otra alteración que presentó el paciente fueron anomalías cardíacas, y precisamente se han identificado varios genes localizados en 9q involucrados en la cardiogénesis (INVS, TMOD1, TGFBR1, KLF4, IPPK, PTCH1, BARX1, MEGF9, y S1PR3)13.
El asesoramiento genético en estos casos es de vital importancia para orientar a los padres sobre posibles causas y explicar sobre la condición genética, su manejo y establecer pautas de seguimiento para hacer prevención terciaria. Por otro lado, en estos casos se debe realizar cariotipo a ambos padres para establecer el estado de portador de alguna aberración cromosómica relacionada con el cromosoma 9 o de efectos intercromosómicos en busca de estudiar a otros hijos y/o realizar planificación familiar.
REFERENCIAS
1. Ioan D., Moiceanu M., Zamfirescu A., Popescu V. Trisomia Pentru Bratul scurt al Cromozomului 9 (trisomia 9p), "de novo". Raportarea Unei Paciente de 2 Ani si 9 Luni. Revista Romana de Pediatrie 2009; vol. lviii, N. 2, An [ Links ]
2. Vaglio A, Mechoso B., Quadrelli A, Quadrelli R. Síndrome de trisomía 9p: características clínico evolutivas y citogenéticas. Arch. Pediatr Urug 2007; 78 (2) [ Links ]
3. Georgianne L., Russell S., Thomas P., Jeffrey R. Trisomy 9: Review and Report of Two New Cases. American Journal of Medical Genetics 1995; 56:252-257 [ Links ]
4. Tonni G., Lituania M., Chitayat D, Bonasoni M, Keathing S., Thompson M., Shannon P. Complete trisomy 9 with unusual phenotypic associations: Dandy-Walker malformation, cleft lip, and cleft palate, cardiovascular abnormalities. [ Links ]
5. Manvelyan M., Simonyan I., Hovhannisyan G., Aroutiounian R., Hamid A., Liehr T. A New Case of a Complex Small Supernumerary Marker Chromosome: A Der(9)t(7;9)(p22;q22) due to a Maternal Balanced Rearrangement. J Pediatr Genet 2015;4:199-200 [ Links ]
6. Bruns DA, Campbell E. Twenty-five additional cases of trisomy 9 mosaic: Birth information, medical conditions, and developmental status. Am J Med Genet 2015 Part A 167A:997-1007. [ Links ]
7. Miryounesi M, Dianatpour M, Shadmani Z, Ghafouri-Fard S. Report of a Case with Trisomy 9 Mosaicism. Iran J Med Sci. 2016;41(3):249-252. [ Links ]
8. Zhou Y., Zhang C., Zhai J., Li T., Wu Q., Li W., Li N., Jun li J., Huang Y, Cui Y., Xia J. A patient with unusual features and a 69.5 Mb duplication from a de novo extra der (9): A case report. Rev Mol Med Rep 2015;12: 155-158 [ Links ]
9. Ma J., Cram D., Zhang J., Shang L., Yang H., Pan H. Birth of a child with trisomy 9 mosaicism syndrome associated with paternal isodisomy 9: case of a positive noninvasive prenatal test result unconfirmed by invasive prenatal diagnosis. Rev Molecular Cytogenetics 2015; 8:44 [ Links ]
10. Sanchez Zahonero J, Mosaicismo de trisomía 9: caso de larga supervivencia. An Peddiatr (Barc), 2008: 68(3):273-6 [ Links ]
11. San Román M., Herranz L., Tejerina A., Arteaga R., Manjón-Cabeza A., López F. Trisomía 9p. An Pediatr (Barc) 2004;61(4):336-9. [ Links ]
12. Santana E., Tamayo V., Pérez G., Guerra V., López A. Manifestaciones fenotípicas de un caso con sospecha clínica de trisomía parcial del cromosoma 9p. Cuba. 2015; Vol.17, No. 1 ISSN 1608 - 8921 [ Links ]
13. Inostroza A., Navarro H., Paublo M., Muñoz H., Hernández A., Catalán J., Sanz P., Puig P. Diagnóstico y Manejo Perinatal de Trisomia 9. Rev Chil Obstet Ginecol 2002; 67(3) [ Links ]
14. Sepulveda W., Wimalasundera C., Taylor M., Blunt S., Bes C., De La Fuente S. Prenatal ultrasound findings in complete trisomy 9. Ultrasound Obstet Gynecol 2003; 22:479-483 [ Links ]
15. Rodríguez S., Monjagata N., Ascurra M. Trisomía parcial del cromosoma 9. Reporte de un caso. Mem. Inst. Investig. Cienc. Salud, 2005;Vol. 3(1) [ Links ]

