











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkCuadernos Hospital de Clínicas
versión impresa ISSN 1562-6776
Cuad. - Hosp. Clín. vol.58 no.1 La Paz 2017
ACTUALIZACIÓN
Algoritmo diagnostico para síndromes genéticos con hipotonía congénita. Parte II
Diagnostic algorithm for genetics syndromes with congenital hypotonia. Part II
Dra. Valeria Aillón López (1), Dra. Beatriz Luna Barrón (2), Dr. Gonzalo Taboada López (3)
(1) Docente Investigador. Instituto de Genética. Universidad Mayor De San Andrés.(2) Docente Investigador. Instituto de Genética. Universidad Mayor De San Andrés.
(3) Docente Investigador Emérito. Instituto de Genética. Universidad Mayor De San Andrés.
Correspondencia: flex_w2@hotmail.com
RECIBIDO: 24/11/2015 ACEPTADO: 01/12/2016
INTRODUCCIÓN
La Hipotonía congénita es un problema común en las unidades de neonatología y de pediatría, así como un motivo frecuente de consulta para el neurólogo pediatra y el genetista, ya que es una de las primeras manifestaciones de los cuadros sindromáticos genéticos y/o hereditarios.1 Al definirse la misma como la disminución del tono muscular en forma generalizada o focal, que habitualmente se asocia a déficit psicomotor1 en la mayoría de los lactantes, la hipotonía se nota a menudo durante o poco después del nacimiento.2 La evaluación del tono muscular es muy subjetiva y depende de una serie de variables, la principal es la experiencia del examinador y la voluntad de llevar a cabo las evaluaciones cuidadosamente. La mayoría de los bebés hipotónicos demuestran una postura característica de plena abducción y rotación externa de las piernas, así como una extensión flácida de los brazos.2
Los avances sin precedentes en la tecnología genética (molecular y citogenética) en las dos últimas décadas llevan un impacto cada vez mayor en nuestra capacidad de proporcionar un diagnóstico específico a nivel molecular para los trastornos genéticos. Por ello la valoración de un síndrome hipotónico exige una evaluación multidisciplinaria: un genetista, un neurólogo pediatra, un especialista en trastornos metabólicos y un neuroradiólogo pediatra entre otros, para poder aplicar con eficacia los avances en el diagnóstico de laboratorio y de imagen.2
En un estudio retrospectivo realizado por Panjan en Ljubljana en Eslovenia, se demuestra que a través de la realización de un algoritmo diagnóstico se puede discriminar la etiología de la hipotonía, es así que la anamnesis y el examen clínico aportaron con 50% del diagnóstico, seguido de un 13% de las técnicas de neuroimagen, la base de datos del Oxford Medical Databases (OMD) aportó con 9%, técnicas de citogenética clásica y molecular con el 6.5%, pruebas bioquímicas con 6%, investigaciones específicas del músculo y nervio con 6% y un restante de exámenes específicos aportaron 6% para el diagnóstico del niño hipotónico.3
ESTUDIOS COMPLEMENTARIOS EN LA HIPOTONÍA CONGÉNITA
Diferenciar las probables causas de hipotonía es importante para evitar someter a los infantes a pruebas diagnósticas invasivas innecesarias, como la biopsia muscular, si la etiología subyacente es probable que sea de origen central en lugar de periférico.4 A pesar de los avances en el diagnóstico electrofisiológico, las pruebas de neuroimagen, y las pruebas moleculares y genéticas, la identificación de la causa de la HC en los niños sigue siendo difícil, excepto en las condiciones más comunes y ampliamente reconocidas, como el síndrome de Down. Así por ejemplo, para los pacientes con hipotonía central, las investigaciones iniciales recomendadas son: cariotipo y estudios de neuroimagen: tomografía computarizada (TC) / resonancia magnética (MRI).5
Estudios de laboratorio:
a) Pruebas biológicas citogenéticas y moleculares: El análisis de citogenética en individuos con diversas anomalías congénitas, características dismórficas, Retraso General del Desarrollo y/o Discapacidad Intelectual se considera generalmente como un componente esencial en el proceso de evaluación; el cariotipo es un estudio básico en genética médica, el proceso se rige según protocolo convencional y se analiza de acuerdo con las directrices del Sistema Internacional de Nomenclatura Citogenética Humanos (ISCN 2013). Es así que el síndrome de Down es la anomalía cromosómica más frecuente6
Las microdeleciones y microduplicaciones presentes en síndromes genéticos por aneusomias segmentarias (Síndrome de Prader Willy, Síndrome Mc Phelan Dermid) pueden hacerse evidentes por medio de citogenética molecular, con el uso de múltiples sondas subteloméricas con Hibridación Fluorescente in situ (FISH), o la técnica de Amplificación de Sondas por Ligación Múltiple (MLPA) en su variante metilación sensible, esta última permite incluso esclarecer 2 de los mecanismos de producción del síndrome de Prader Willy siendo útil para el asesoramiento genético y la orientación hacia otros estudios (búsqueda de disomía uniparental por STR's o estudio de mutación específica) . El diagnóstico molecular proporciona la ventaja de la velocidad y la especificidad diagnóstica sin ser invasiva.5 Pruebas de diagnóstico basados en el ADN establecen el diagnóstico de Atrofia Muscular Espinal,5 así por ejemplo, en la mayoría de los bebés con AME tipo I (enfermedad de Werdnig-Hoffman) la presentación clínica es típica y el diagnóstico puede ser fácilmente confirmado por análisis de ADN realizando PCR para las deleciones de los exones 7 y 8 del gen (5q11-13), obviando así la necesidad de EMG o biopsia muscular.7
Finalmente, el análisis de mutación directa para la deleción de las neuronas motoras (SMN) y la distrofia miotónica (detección de repetición de triplete) deben llevarse a cabo de acuerdo a la presentación clínica.5
b) Errores Innatos del Metabolismo. (EIM). Si la evaluación clínica sugiere afectación multisistémica compleja (convulsiones, letargía, rechazo a la alimentación) es útil sospechar de este grupo de patologías3. Los defectos bioquímicos pertenecen a una de tres categorías: las encefalopatías tóxicas (acumulación de metabolitos tóxicos), encefalopatías de energía deficiente (errores congénitos que afectan la producción o utilización de la energía) y trastornos que afectan el procesamiento intracelular de moléculas complejas. Por ello la realización de un tamiz neonatal ampliado es de vital importancia en todo recién nacido, por otra parta el laboratorio básico en EIM incluye: Medición de amonio sérico (defectos del ciclo de la urea, acidemias orgánicas, trastornos de la oxidación de ácidos grasos) y lactato (trastornos del metabolismo de carbohidratos, enfermedad mitocondrial) en la sangre y otros fluidos corporales (orina y LCR). El análisis cuantitativo de aminoácidos en sangre y orina para aminoacidopatías, perfiles de ácidos orgánicos y acilcarnitinas en la sangre utilizando espectrometría de masas en tándem (MS-MS) para acidemias orgánicas y/o defectos de la oxidación de ácidos grasos. Los ensayos de ácidos grasos de cadena muy larga (VLCFA) en el plasma para trastornos peroxisomales, electroforesis isoinmune de transferrina baja en los trastornos de la glicosilación y 7- dehidrocolesterol: elevada en el síndrome de Smith-Lemli-Opitz (SLO).2 La medición de ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; CK: creatincinasa; LDH: lactato deshidrogenasa son indicadores sensibles, pero no específicos de la enfermedad de Pompe de inicio tardío, ya que se puede observar en el 95% de los afectados; el análisis de la actividad α-glucosidasa es imprescindible en el diagnóstico de la enfermedad de Pompe de inicio tardío mediante el método de la muestra en sangre seca, este hallazgo debe confirmarse con un estudio enzimático en muestra líquida en linfocitos aislados.8 Estas pruebas permiten la detección de metabolitos anormales que son marcadores bioquímicos de trastornos metabólicos conocidos. La consulta con un especialista en genética, es a menudo muy útil en la priorización de las investigaciones.5
c) Enzimas musculares (creatina fosfoquinasa) rara vez son útiles en un niño con hipotonía, con la excepción de los trastornos musculares, donde se elevan los valores de creatina quinasa (CPK) y en algunas formas de las miopatías congénitas.
Estudios de Gabinete
a) Estudios de neuroimagen: Estudios como Tomografía Computarizada (TC)/ Resonancia Magnética Nuclear (RMN) del cerebro son útiles en la identificación de malformaciones estructurales, defectos de migración neuronal (por ejemplo lisencefalia), características de la señal alteradas de la sustancia blanca (por ejemplo, la deficiencia de laminina). Anormalidades de señal en los ganglios basales (por ejemplo, citopatías mitocondriales), así como la detección de anomalías del cerebelo, "signo del diente molar 'en el síndrome de Joubert y agenesia del cuerpo calloso en la deficiencia del complejo piruvato deshidrogenasa y otros trastornos metabólicos son hallazgos que pueden ser patognomónicos para trastornos específicos2 En los pacientes afectados por la enfermedad de Pompe, no se observa una distribución específica de la atrofia muscular y la infiltración grasa, pero, empleando un protocolo de resonancia magnética de cuerpo entero, se puede obtener un patrón sugerente de esta enfermedad.8
b) Estudios electrofisiológicos: En el caso de hipotonía derivada de una lesión en la unidad motora inferior, pruebas invasivas se hacen necesarias, tales como la conducción nerviosa y electromiografía (EMG). Para el estudio de Atrofia Espinomuscular (SMA) la EMG es un estudio muy útil. Hallazgos miopáticos incluyen potenciales de baja amplitud de acción muscular compuesto (CMAPs) y pequeños potenciales de unidad motora polifásicos que son reclutados rápidamente. Estudios de conducción nerviosa y EMG también son útiles en la investigación de las neuropatías sensoriales y motoras hereditarias y en la distinción de los trastornos axonales de las condiciones desmielinizantes.4 Los exámenes electrofisiológicos en los pacientes con enfermedad de Pompe suelen mostrar estudios de conducción nerviosa normales, pero el electromiograma puede presentar un patrón miopático en la musculatura proximal y signos de irritabilidad de membrana en forma de descargas miotónicas, preferentemente en la musculatura paravertebral.8
c) Biopsia muscular: Se debe considerar en el diagnóstico de sospecha de miopatías y distrofias musculares, incluso si los estudios electrofisiológicos son normales. Técnicas de inmunohistoquímica que utilizan anticuerpos contra las proteínas musculares son especialmente útiles en el diagnóstico de las distrofias musculares congénitas (CMD), miopatías y mitocondriopatías.5 En la biopsia muscular de pacientes con Enfermedad de Pompe se observa una miopatía vacuolar con depósito de glucógeno, aunque puede ser normal o inespecífica en el 30% de los casos.8
ALGORITMO DIAGNÓSTICO
Para llegar al diagnóstico definitivo del niño hipotónico una vez realizada la revisión bibliográfica correspondiente al tema, se propone el siguiente algoritmo diagnóstico (ver figura 1).
Cuando se tiene un paciente hipotónico, primeramente pasará por evaluación neurológica para establecer el origen: central o periférico, de acuerdo a las características de cada una. Sí, se encuentra patología neurológica pura, se realizará el seguimiento por neurología; caso contrario, pasa a consulta por genética médica, para estudiar Síndromes Genéticos Asociados, dentro de estos, se buscarán de origen central:
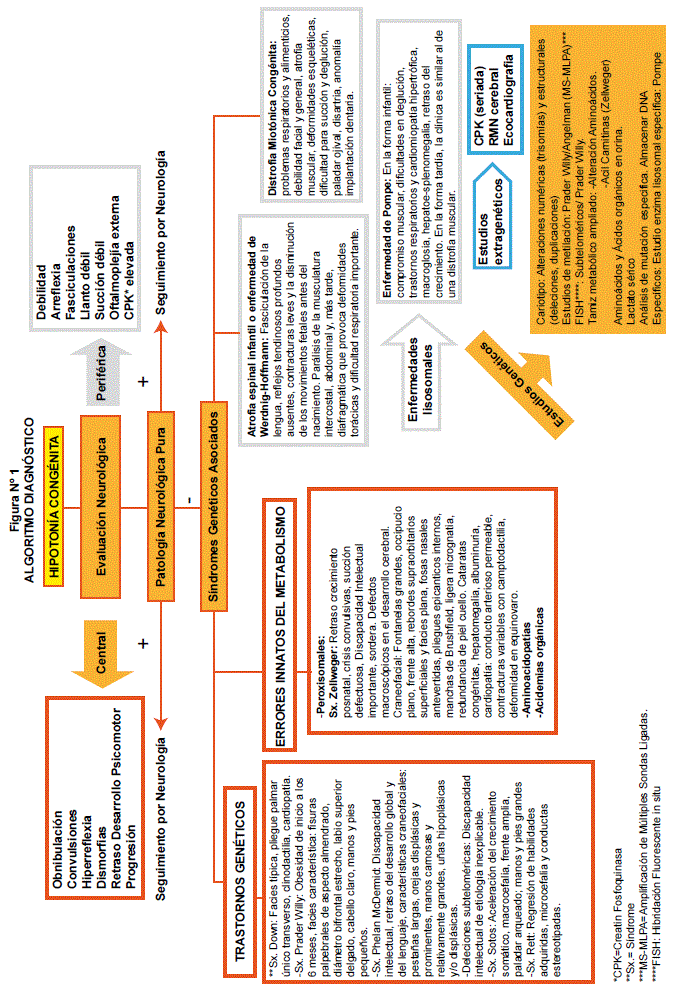
Trastornos Cromosómicos tales como: Sx. Down, Sx. Prader-Willy, Sx. Phelan McDermid, cada uno con las características ya descritas. En Errores Innatos del Metabolismo tenemos: Síndromes Peroxisomales: Sx. Zellweger; Aminoacidopatías y Acidemias orgánicas. Por otra parte, de origen periférico podemos encontrar: Enfermedades lisosomales: Enfermedad de Pompe; Atrofia espinal infantil o enfermedad de Werdnig-Hoffmann, Distrofia Miotónica Congénita. Para confirmar su etiología, se procederá a realizar estudios genéticos como: Cariotipo convencional, citogenética molecular con estudios de metilación: Prader Willy. FISH: Subteloméricos/ Prader Willy. Tamiz metabólico ampliado: Para Alteración de Aminoácidos, Acil Carnitinas (Zellwegwer). Aminoácidos y Ácidos orgánicos en orina. Lactato sérico. Análisis de mutación específica: Almacenar DNA. Específicos: Estudio enzima lisosomal específica: Pompe. Como estudios extragénicos: CPK (seriada), RMN cerebral y Ecocardiografia.
DISCUSIÓN
La evaluación de la HC debe incluir una genealogía detallada y documentación de los factores de riesgo prenatales. En la exploración física es de importancia establecer la diferencia entre un origen central y un periférico, buscar dismorfias faciales y/o corporales, anomalías congénitas y datos de afección multisistémica. Tal como afirma en el estudio retrospectivo que realizó Paro Panjan, el 50% para un diagnóstico específico de hipotonía se debe a una correcta anamnesis y exploración física (3). Para completar el panorama clínico, los estudios laboratoriales terminarán de confirmar la sospecha diagnóstica previa. (7) Se sugiere un enfoque sistemático basado en las pruebas actualmente utilizadas en la evaluación de los niños con hipotonía, los detalles clínicos y pruebas de diagnóstico pertinentes que se analizan en conjunto con trastornos específicos (2, 3,4). Es así que, teniendo una aproximación clínica con el soporte clínico y laboratorial, los casos de Hipotonía Congénita sin determinar se verán más fácilmente evaluados de acuerdo a los pasos previos y en última instancia el objetivo de contar con un diagnóstico preciso es poder brindar una orientación terapéutica dirigida a cada caso en particular (5,8).
CONCLUSIÓN
De esta manera, a través de una guía o algoritmo diagnóstico el estudio de la Hipotonía Congénita se facilita de mejor manera, no obstante, la diversidad y complejidad de este tipo de patologías justifica el abordaje multidisciplinario.
REFERENCIAS
1. Aillón López Valeria, Luna Barrón Beatriz y Taboada López Gonzalo. Hipotonia Congénita Y Síndromes Genéticos. Revista Cuadernos. 2016 (in press). [ Links ]
2. Prasad A. and Prasad C. Genetic evaluation of the floppy infant. Seminars in Fetal & Neonatal Medicine 16 (2011)99-108 [ Links ]
3. Darja Paro-Panjan, David Neubauer. Congenital Hypotonia: Is There an Algorithm. J Child Neurol. 2004 Jun; 19(6):439-42. [ Links ]
4. Harris Susan R. Congenital hypotonia: clinical and developmental assessment. Dev Med Child Neurol. 2008 Dec;50(12):889-92 [ Links ]
5. Ibarra Lúzar J., Estévez Poy P. Distrofia miotónica congénita. Hallazgos clínicos, electrofisiológicos y genéticos de nuestra casuística. Elsevier. Vol. 43. Núm. 04. Julio - Agosto 2009 [ Links ]
6. Uwineza Annette, Hitayezu Janvier, jamar Mauricette et al. Cytogenetic Studies of Rwandan Pediatric Patients Presenting with Global Developmental Delay, Intellectual Disability andlor Multiple Congenital Anomalies. Journal of Tropical Pediatrics, 2016, 62, 38-45 [ Links ]
7. Darras BT, Jones HR. Diagnosis of pediatric neuromuscular disorders in the era of DNA analysis. Pediatr Neurol. 2000 ct; 23 (4):289-300. [ Links ]
8. Romero Miguel, Barrot Emilia, Lorite Juan Bautista, et al. Guía clínica de la Enfermedad de Pompe de inicio tardío. Rev Neurol 2012; 54 (8): 497-507 [ Links ]

