











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkCuadernos Hospital de Clínicas
versión impresa ISSN 1562-6776
Cuad. - Hosp. Clín. vol.57 no.1 La Paz 2016
CASOS CLÍNICOS
Mielolipoma adrenal
Reporte de caso y revisión de la literatura
Adrenal myelolipoma
Case reportand review of literature
Dr. Carlos Dencel Peláez Pacheco1, Dr. Alfredo Cari Siles2, Dra. Viviana Raquel Barrón Mondaca3.
1Cirujano General, Cirujano Oncólogo. Servicio de Oncología de la Caja Nacional de Salud. La Paz - Bolivia.
2Cirujano General. Servicio de Oncología de La Caja Nacional de Salud. La Paz - Bolivia.
3Médica Anatomopatóloga. Servicio de Patología de La Caja Nacional de Salud. La Paz - Bolivia.
Correspondencia: Dr.Pelaez@biociencias.org
Resumen
El mielolipoma adrenal es un tumor benigno de etiología desconocida y de presentación infrecuente. Generalmente asintomático y poco asociado a producción hormonal. Conlleva dificultades diagnósticas y terapéuticas de acuerdo a cada caso en particular.
Se presenta el caso de un mielolipoma adrenal derecho en mujer adulta eritrocitica, obesa, hipertensa, con síndrome adherencial severo secundario a múltiples cirugías abdominales. Planteó dificultad diagnóstica y terapéutica por asociación a elevaciones de presión arterial y posibilidad de tumor funcionante.
Palabras claves: mielolipoma, adrenal, caso clínico, tratamiento quirúrgico.
INTRODUCCIÓN
El mielolipoma es un tumor benigno de rara frecuencia de presentación 0,03 a 0,8%. Si tomamos la incidencia en tumores adrenales primarios su presentación es del 5 a 7%.1, 2, 3
La etiología es desconocida sin embargo se considera a un tumor compuesto por adipocitos maduros y tejido hematopoyético con metaplasia retículo endotelial de los capilares sanguíneos en la glándula adrenal que responde a infección, trauma o necrosis.2
Generalmente es unilateral, lado derecho, raramente bilateral, la localización extra adrenal es infrecuente y ha sido descrita en retroperitoneo, tórax y pelvis.4
Es más prevalente en la quinta a séptima decada de vida sin predilección de sexo. Está relacionado a obesidad hipertensión y diabetes.1, 2, 5
Generalmente cursan asintomáticos, se ha establecido la presencia de 1% en autopsias.4 Muchos han sido descubiertos accidentalmente.5 Cuando es sintomático predominan el dolor y el sangrado. Los síntomas más serios descritos fueron hematuria, hemorragia e hipertensión renovascular.7 Comúnmente no son hormonalmente funcionantes.2 Tumores mayores de 4 cm se asocian a mayores complicaciones.
El diagnóstico se sospecha en la ecografía y debe complementarse con tomografía buscando tumores adrenales con densidad grasa (< 30 UH) 1,4 Puede utilizarse la resonancia magnética donde la presentación es similar en T1 y T2.
En caso de duda diagnóstica se recomienda la biopsia guiada por ecografía.4, 6
Dentro los diagnósticos diferenciales deben tenerse en cuenta los adenomas, angiomiolipoma renal, lipoma retroperitoneal, liposarcoma, feocromocitomas, ganglioneuroma, carcinomas, o metástasis.5, 7
El tratamiento es quirúrgico si el tumor es sintomático, hormonalmente activo y/o mayor a 3,5 cm 1, 8, 9. La resección puede realizarse en tumores menores a 7 cm por vía laparoscópica. Cirugía abierta en caso de tumores de mayor diámetro.8,9 Ya se inició cirugía robótica en tumores menores de 7 cm con buenos resultados.10
El pronóstico es excelente ya que no presenta recidiva.
En Bolivia no se han publicado casos respecto a mielolipomas adrenales. Su presentación infrecuente implica dificultades en la confirmación diagnóstica y en la toma de decisiones como el caso que se presenta a continuación.
REPORTE DE CASO
Mujer de 56 años natural de la ciudad La Paz y residente en la ciudad de El Alto, Bolivia; profesora, sin hábitos tóxicos. Antecedentes de eritrocitosis de altura, hipertensión arterial de 6 años de evolución en tratamiento con antagonistas de la angiotensina II; rosácea, hipertrigliceridemia, tuberculosis pleural con tratamiento concluido, adenoma tiroideo asintomatico, histe recto mía, dos cesáreas, colecistectomía. No antecedentes familiares de cáncer.
Acude a primera consulta por cuadro clínico de 7 meses de evolución con disuria, dolor abdominal inespecífico, alzas térmicas dudosas, no cualificadas y malestar general. Ante hallazgo tomográfico de tumor adrenal derecho fue derivado al servicio de Oncología.
Al examen físico buen estado general, obesa tipo II (IMC: 37), facies eritrocitica, signos vitales normales, tendencia a hipertensión PA: 130/80. No adenopatías.
Cardiopulmonar sin datos llamativos, abdomen con múltiples cicatrices anteriores y abundante tejido celular subcutáneo que no permite palpación de tumores, No ascitis, No signos peritoneales. Genitourinario, extremidades, neurológico sin particularidades.
EXAMENES COMPLEMENTARIOS
LABORATORIO: Leucograma, glucemia, creatinina, NUS, electrolitos, calcemia, perfil hepático, coagulación, proteinas totales, albumina, ácido úrico VES; dentro de parámetros normales. Hb: 16,5 mg.
Cortisol: normal a la toma de 8 a.m. 80 nmol/L. Catecolaminas en orina de 24 horas: levemente incrementado con 711 mcg/24 hrs valor laboratorial de referencia (200 a 700).
Rx PA de tórax: no metástasis, no derrame pleural, leve incremento de trama vascular, no cardiomegalia.
TAC abdomen y pelvis: tumor suprarrenal derecho heterogéneo redondeado, hipodenso con coeficiente de atenuación -38 UH grasa. Mide 66 mm. No adenopatías, no líquido libre, no otros tumores. Imágenes 1 y 2.
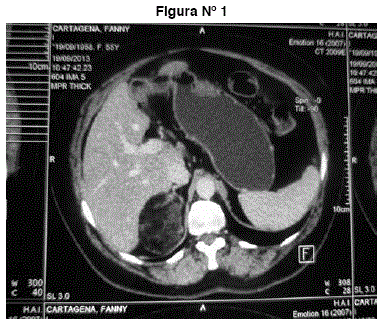
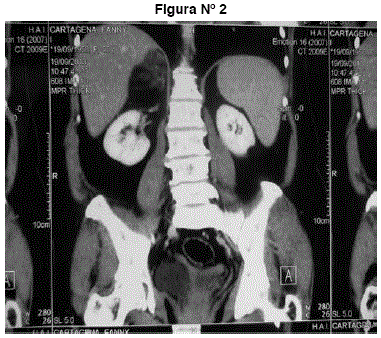
Figuras N° 1 y N° 2. Tumor suprarrenal derecho heterogéneo redondeado, hipodenso con coeficiente de atenuación -38 UH grasa. Mide 66 mm. No adenopatías, no líquido libre, no otros tumores.
Fue valorada por, Cardiología, Medicina Interna, Hematología, Anestesiología y Terapia Intensiva. En pre operatorio fue sometida a sangría de 500 mL más reposición por eritrocitosis.
En sala de operaciones presentó hipertensión arterial con valor máximo de 170/110 PAM: 134; por lo que se reprogramo la cirugía y se preparó pos operatorio en Terapia Intensiva ante posibilidad de tumor funcionante.
Fue sometida a laparotomía evidenciando bridas y adherencias epiploparietales, epiplointestinales e interasas en todo el abdomen a predominio suhepatico. Tumorde 7 cm en glándula suprrarenal derecha, superficie lisa, multilobulado, blando, de apariencia lipomatoso. No liquido libre, no adenopatías, no otras tumoraciones.
Se procedió a liberación de adherencias, adrenalectomia derecha. El procedimiento fue dificultoso por síndrome adherencial. Durante el procedimiento no presento variaciones importantes en la presión arterial.
El pos operatorio fue controlado por dos días en terapia intensiva sin presentar eventualidades, es externada al 5 día pos operatorio.
El reporte de histopatología de la pieza operatoria informa: masa ovoidea de tejido blando, bien encapsulado, pardo violáceo que mide 8,7 x 6,6 x 4 cm y pesa 90 g, presenta en uno de sus extremos un área pardoamarillenta que mide 4 x 3,5 x 3,5 cm. Al corte superficies de sección heterogéneas, muestra áreas hemorrágicas entremezcladas con áreas pardo grisáceas. Imagen 3. Concluye: adrenalectomia derecha con mielolipoma capsulado intacto.
DISCUSIÓN
Desde su descripción en 1905 por Gierke 4,6 el mielolipoma adrenal a sufrido cambios en su presentación, diagnóstico y en su tratamiento.
Aún se desconoce su etiología, y dentro las varias hipótesis la más aceptada se refiere a que los elementos hematopoyéticos y grasos pueden ser derivados de una célula progenitora común 5, 11, 12
En cuanto a la presentación de acuerdo a sexo, no se encuentra del todo definido. Muchos artículos señalan mayor presentación en mujeres y otros concluyen sin predilección.1, 2, 5, 7,12
Como en nuestro caso, se ha descrito relación con obesidad, hipertensión, diabetes.5Aquello conlleva mayor dificultad diagnóstica y terapeútica.
La mayoría son asintomáticos, por eso pueden pasar desapercibidos, incluso ser enmascarados hasta llegar a complicaciones extremas como ruptura espontánea, hemorragia, shock y muerte.13
Muchos de ellos son detectados incidentalmente, varios de ellos por ecografía, sin embargo este método no es el mejor por su baja sensibilidad y especificidad. Actualmente la tomografía es el método de elección debido a su alto poder en la determinación de densidades imagenológicas que presenta este tipo de tumoración.5,11
Los diagnósticos diferenciales son varios, ya que existen muchos tipos de tumores localizados en la glándula adrenal, pudiendo ser benignos o malignos. Cualquier diagnóstico diferencial puede necesitar otro tipo de decisiones terapeúticas.5,14
Dentro de los diagnósticos más dificultosos, conlleva a determinar si el tumor es o no hormonalmente funcionante. O si es que se trata de otro tipo de tumor funcionante como el feocromocitoma, Aquello sucedió en nuestro caso.
La dificultad en caso de un tumor funcionante radica en que durante la extirpación tumoral se pueden desencadenar trastornos hormonales con sobreproducción de catecolaminas por manipulación. Teniendo como consecuencia crisis hipertensivas y riesgo alto de muerte.1, 2, 3 La preparación pre operatoria será fundamental para evitar cualquier eventualidad, en nuestro caso con laboratorios respecto a la detección de catecolaminas en orina de 24 horas, cortisol en sangre, revisión de estudios de imagen.
No realizamos la biopsia preoperatoria ni preparación con beta bloqueantes; ya que los estudios mencionados alejaban la posibilidad de un tumor funcionante y preferimos evitar el riesgo de ruptura tumoral, sangrado o incluso crisis hipertensiva durante la biopsia. Sin embargo ante la posibilidad; nos preparamos para control trans y pos operatorio, en caso de una "tormenta hipertensiva".
Los mielolipomas extraadrenales son infrecuentes, tampoco se ha demostrado su relación y frecuencia de presentación con mielolipomas adrenales. Cada caso deberá ser evaluado por el equipo médico y descartar la asociación si así se considere.15
La cirugía ha tenido la mayor evolución en el tratamiento de mielolipomas adrenales. Las indicaciones son más precisas y tratan de evitar las complicaciones que incrementan en tumores mayores a 3,5 cm.8,9,10,16 Se ha incluido dentro las complicaciones la malignización en tumores de gran tamaño.17 Esta aceptado el tratamiento por via laparóscopica en tumores menores de 7 cm. En nuestro caso un síndrome adherencial severo secundario a cirugías abdominopelvicas previas, contraindico cualquier intento por laparoscopia, y de echo requirió un esfuerzo mayor.
El mielolipoma de mayor tamaño reportado fue de 31 x 24,5 cm con 6 Kg.5
El diagnóstico, preparación preoperatoria, toma de decisiones, tratamiento y manejo pos operatorio requirió un adecuado manejo multidisciplinario, el cual servirá de referencia para el manejo de futuras presentaciones.
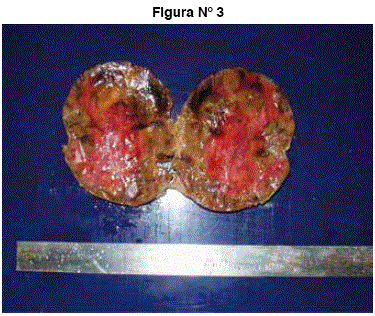
Figura N° 3: Masa ovoidea de tejido blando, bien encapsulado, pardo violáceo que mide 8,7 x 6,6 x 4 cm y pesa 90 g, presenta en uno de sus extremos un área pardoamarillenta que mide 4 x 3,5 x 3,5 cm. Al corte superficies de sección heterogéneas, muestra áreas hemorrágicas entremezcladas con aéreas pardo grisáceas.
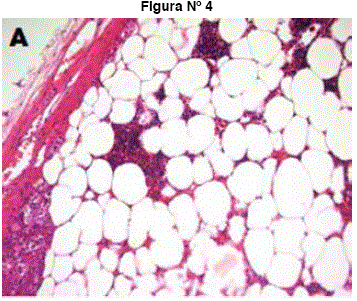
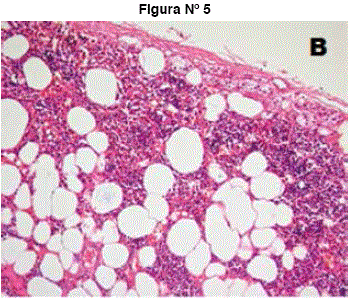
Figuras N° 4 y N° 5 Histología muestra: Islas de elementos hematopoyéticos entremezclados con adipocitos maduros, en extremo inferior izquierdo corteza adrenal. (A). Elementos eritropoyéticos normales tales como eritroblastos, células linfoides, megacariocitos inmaduros y células grasa.(B)
REFERENCIAS
1. Sippel, O.; Sue O'Dorisio, M.; VinikA. I.: The North American Neuroendocrine Tumor Society Consensus Guideline for the Diagnosis and Management of Neuroendocrine Tumors, Pheochromocytoma, Paraganglioma, and Medullary Thyroid Cancer. Pancreas Volume 39, Number 6, August 2010. [ Links ]
2. Mansur, J. D.; Espada, J. D.; Mandarino, A.: Paragangliomavesical. Presentación de un caso y suresolución. Rev. Arg. de Urol. Vol. 69 (2) 2004 118-122. [ Links ]
3. Dundr, P.; Dudorkinová, D.; Povy's, C.: Pigmented Composite Paraganglioma - Ganglioneuroma of the Urinary Bladder. Pathol. Res. Pract. 199: 765-769 (2003). [ Links ]
4. Deng, J.H.; Li, H. Z.; Zhang, Y.: Functional paraganglioma sof the urinary bladder: a report of 9 cases. Chin J Cancer2010;29(8):729-734. [ Links ]
5. Diaz- Nuñez, J. R.; Hernandez - Martinez, G.; Rodriguez - Montes C.: Paraganglioma vesical. Rev MexUrol 2011 ;71(3):172-175). [ Links ]
6. Correa, JJ.; Velez, A.; Hessen, M.: Paraganglioma vesical en adulto. Reportede un caso. Urol. Colomb. Vol XX, No. 3:pp. 78-81, 2011. [ Links ]
7. Davis, CJ.: Tumours of the urinary. World Health Organization Classification of Rumours IARC; 2004: 136-137. [ Links ]
8. Salgado, G.; Marin, D.; Espinoza, K.: Paragangliomas: Metodos de imagen y correlación histopatológica. Anales de Radiología México 2009;4:307-317. [ Links ]
9. Xu, DF.; Chen, M.; Liu, YS.; Gao, Y.; Cui, XG.: Nonfunctional paraganglioma of the urinary bladder: a case report. J Med Case Reports 2010 Jul 19; 4:216. [ Links ]
10. Villaquirána, C.; García-Perdomob, H.; Ramírez, G.: Paraganglioma vesical: una entidad poco frecuente con tratamiento controversial. . RevMexUrol 2013;73(5):267-270. [ Links ]

