











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkCuadernos Hospital de Clínicas
versión impresa ISSN 1562-6776
Cuad. - Hosp. Clín. vol.55 no.2 La Paz 2014
CASOS CLÍNICOS
Presentación de un caso clínico de mucopolisacaridosis Tipo IV, síndrome de Morquio
Presentation of a clinical case of mucopolysaccharidosis type IV, Morquio syndrome
Aguilar, Ximena A (1), Rafael Montaño A (2), Roxana Saunero (3), Álvaro Gálvez L (4)
1 Docente Investigador, Instituto de Genética -UMSA.
2 Tesista de Maestría en Ciencias Biológicas y Biomédicas, Instituto de Genética -UMSA.
3 Pediatra del Hospital Materno Infantil -Caja Nacional de Salud.
4 Auxliar de Investigación, Instituto de Genética -UMSA.
Resumen
Se presenta el caso de un paciente de 16 años de edad, con el diagnostico de mucopolisacaridosis (MPS) tipo IV-A, con una breve revisión teórica del curso y progresión crónica de esta enfermedad multi-sistémica, que se manifiesta con amplia signo sintomatología, hallazgos de laboratorio y anomalías radiológicas. El objetivo es documentar el caso y difundir a la comunidad médica boliviana, la importancia de los errores innatos del metabolismo, consideradas enfermedades "raras", que a criterio nuestro, sufren un sub-diagnóstico debido a las pocas publicaciones científicas sobre el tema en el medio.
Palabras claves:
Síndrome de Morquio, Mucopolisacaridosis IV A, Enfermedad de Morquio, Deficiencia de Galactosamida- 6- Sulfatasa
Abstract
We report the case of a patient 16 years old with a diagnosis of mucopolysaccharidosis (MPS) type IV- A, with a brief theoretical review of chronic course and progression of this multisystem disease, which manifests with extensive signs symptoms, findings are presented, with laboratory and radiological reported abnormalities. The aim is to document the event and communicated to Bolivian medical community, the importance of inborn errors of metabolism, considered "rare" diseases, which in our opinion; suffer a sub- diagnosis because of the few Bolivian scientific publications on the topic.
Keywords:
Morquio Syndrome, MPS IVA Morquio A Disease, Galactosamine-6-Sulfatase Deficiency
INTRODUCCIÓN
La mucopolisacáridosis tipo IV (MSP IV) o síndrome de Morquio (SM), toma su nombre del Dr. Luis Morquio, pediatra uruguayo, quien en 1929 describió la patología en una familia de ascendencia sueca con cuatro niños afectados. Fue descrita el mismo año por el Dr. Brailsforden Inglaterra por lo que se la conoce también como síndrome de Morquio-Brailsford. Se distinguen tres formas de la enfermedad, atendiendo fundamentalmente a diferencias enzimáticas para su diagnóstico debido a que clínicamente son indistinguibles entre ellas. Estas formas son la A, B y C. (3, 4, 5). Esta peculiaridad se debe a que los mucopolisacáridos o glucosaminoglicanos (GAG) son cadenas de disacáridos repetidos, específicos de alto peso molecular, sintetizados por las células del tejido conectivo. La naturaleza de las dos moléculas de azúcar, es la característica diferencial entre los distintos tipos de GAG. La degradación de estas moléculas se da en los lisosomas, por medio de enzimas específicas para el monosacárido terminal de la molécula disacárida, junto con el tipo de enlace que presenta con el azúcar siguiente. Dado que son específicas de monosacárido, y no de disacárido, una enzima puede ser común para el metabolismo de distintos tipos de GAG (1).
Las mucopolisacaridosis (MPS) constituyen un grupo heterogéneo de dolencias hereditarias caracterizadas por la anormalidad del metabolismo de estos GAG, los cuales se acumulan en lisosomas debido a deficiencias enzimáticas lisosomales específicas y genéticas. Estas entidades constituyen alrededor del 32% del total de Errores Innatos del Metabolismo (EIM) (2). Con excepción de la MPS tipo II o Síndrome de Hunter que tiene una herencia ligada al cromosoma X, el resto de MPS son de herencia autosómica recesiva.
El tipo A, también llamado "forma clásica", presenta los cuadros clínicos más severos. Está caracterizadoporfaltatotaldelfuncionamientodela enzima N-acetilgalactosamina-6-sulfato sulfatasa (6-sulfo-N-acetilhexosaminida sulfatasa), debido a mutaciones en el gen GALNS (locus 16q24.3) (3). Se ha asociado también a deficiencia parcial de la enzima neuraminidasa debido a mutaciones en el gen NEU1 (locus 6p21.3). El tipo B (3p21.33 gen GLB1 galactocidasa, Beta-1) resulta de la ausencia de funcionamiento casi total de la enzima beta-galactosidasa (4). En ambos tipos se caracterizan por la eliminación de queratán-sulfato por la orina; determinado por la imposibilidad de las células retículo-endoteliales de degradar este producto de la síntesis normal de sustancia fundamental del tejido conectivo, ocasionando su acúmulo.
El tipo C se caracteriza por la ausencia de este fenómeno, denominándose también Síndrome de Morquio no excretor de queratán-sulfato (5). En este tipo de presentación, no se detectan mucopolisacáridos (MPs) en la orina, ni se observa déficit funcional de galactosamina-6 sulfatasa, ni de beta-galactosidasa en leucocitos y fibroblastos (rasgos que definen los tipos A y B respectivamente) (3,6). Actualmente se cree que es una variedad alélica del tipo A o B, en la que la normal disminución de los niveles urinarios de MPs observados con el pasar de los años, en estos pacientes se vería acelerado, quizá por una actividad residual mínima indetectable de las enzimas implicadas (3,5). Sin embargo, para afirmar esto se requieren todavía mayores estudios.
Sin importar la variedad, todos se manifiestan debido a un acúmulo exagerado de MPs (fundamentalmente queratán-sulfato y condroitín-6-sulfato) en los lisosomas de distintas células del organismo. Parte de la clínica del síndrome se explica justamente por el tipo celular que se acumula. Los niños empiezan a manifestar los rasgos de la enfermedad entre el primer y tercer año de vida. Se describe el descenso de la curva de crecimiento a partir del año de edad. El tronco corto, cuello corto, genu valgo, cifosis, platispondilia, hipoplasia odontoide, prognatismo, dientes pequeños, disostosis múltiple, son signos frecuentemente descritos. Así como las infecciones del tracto respiratorio superior recurrentes y otitis media, además de hernias; Siendo ya todas estas características, evidentes entre los 18 meses y los 2 años de vida (7). Esta inexorable progresión determina el pronóstico tan sombrío a mediano y largo plazo, y mayor número de células afectadas. La expectativa de vida en los casos severos, generalmente de MPS IV-A, no suele exceder los 25 años sobreviniendo la muerte debido a complicaciones cardíacas o respiratorias. Los casos menos graves, comúnmente de MPS IV tipo B o C, pueden exceder los 60 años de vida (3)
A diferencia de la mayoría de los Errores Innatos del Metabolismo (EIM), en esta patología destaca la absoluta indemnidad del desarrollo intelectual a lo largo de la vida. Resulta importante recordar este dato a la hora de diferenciar la MPS IV de otros tipos de MPS, que sí cursan con déficit mental, como el síndrome de Hurler (MPS tipo I) o el síndrome de Hunter (MPS tipo II), (1, 8 ,11) ambas igualmente con evolución y pronóstico grave pero con otro tipo de manifestaciones y complicaciones.
La frecuencia de las enfermedades por depósito en los lisosomas se calcula en 1:5.000 a 1:8.000 r.n.v de manera global (6). Su distribución es mundial, a pesar de la mayor prevalencia de ciertas enfermedades concretas en determinados grupos de poblaciones (ej.Judíos askenazíes con enfermedad de Gaucher y Tay Sachs(6)). Concretamente, las frecuencias observadas del SM se calculan en 1:210.000 r.n.v. (9), pese a que los reportes varían entre países existiendo reportes que van desde 1:200,000 a 1:600,000. Datos canadienses recientes mencionan una frecuencia de 1:216,412 r.n.v., mientras que datos australianos hablan de 1:640,000 r.n.v. (6).
CASO CLÍNICO:
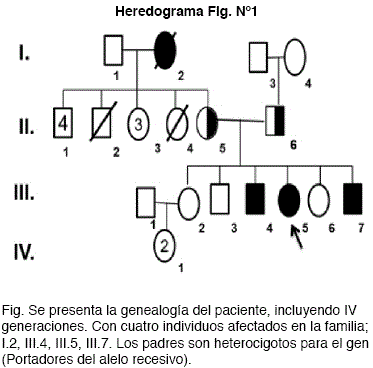
Presentamos el caso de un paciente de 16 años de edad, sexo masculino, procedente y residente de Huachía, (Apolo-Provincia Franz Tamayo de La Paz, Bolivia). Padre de 45 años, madre de 38 años. Como antecedentes familiares referidos: La abuela materna (Fallecida (I.2), tenía características físicas similares; al igual que un hermano (III.5) y una hermana (III.7).
Dentro de los antecedentes gineco-obstétricos de la madre se refiere gestas 6 y partos 6, todos nacidos vivos. El paciente producto de la 3ra gestación, a término, sin intercurrencias. Parto eutócico, domiciliario con presencia de llanto al nacer sin necesidad de asistencia neonatal.
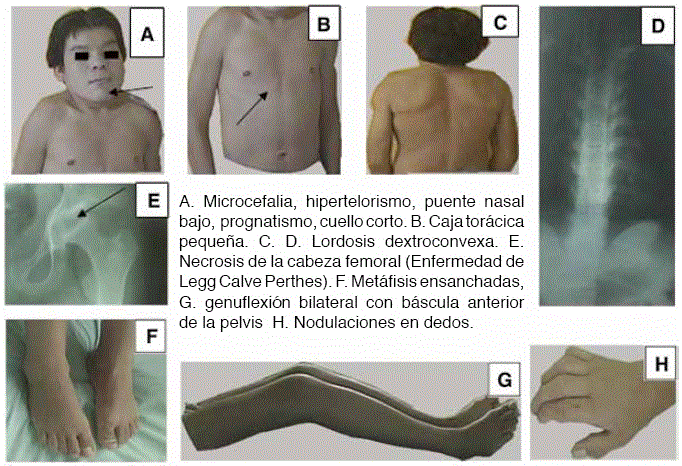
Al examen físico segmentario se observa normocefalia, con pabellones auriculares de implantación normal y ojos con hipertelorismo verdadero (por encima del percentil 97%) (10). Nariz con puente nasal bajo y estenosis anterior de coanas. En maxilar se observa prognatismo, paladar alto, cuello corto, caja torácica pequeña. Escoliosis toracolumbar. Porestas malformaciones se hace imposible tallar adecuadamente al paciente dado que le es imposible pararse completamente erguido.
Destaca en rodillas, genuflexión bilateral y genuvalgun, con báscula anterior de la pelvis, hiperlordosis compensatoria, muñecas con limitación en la dorsiflexión y desviación cubital, nodulaciones en articulaciones interfalángicas de ambas manos, articulaciones interfalángicas en semiflexión limitada.
Exámenes Complementarios:
Ecocardiografía: Hipertrofia septal interventricular asimétrica, dilatación de la raíz aortica, displasia de válvula de la aorta.
Radiología: Las imágenes radiológicas de columna mostraron en la radiología: escoliosis lumbar, dextroconvexa, deformidad de cuerpos vertebrales lumbares, listesis anterior de L4 sobre L5, enfermedad de Legg-Calve-Perthes. En cadera presenta necrosis de ambas cabezas femorales.
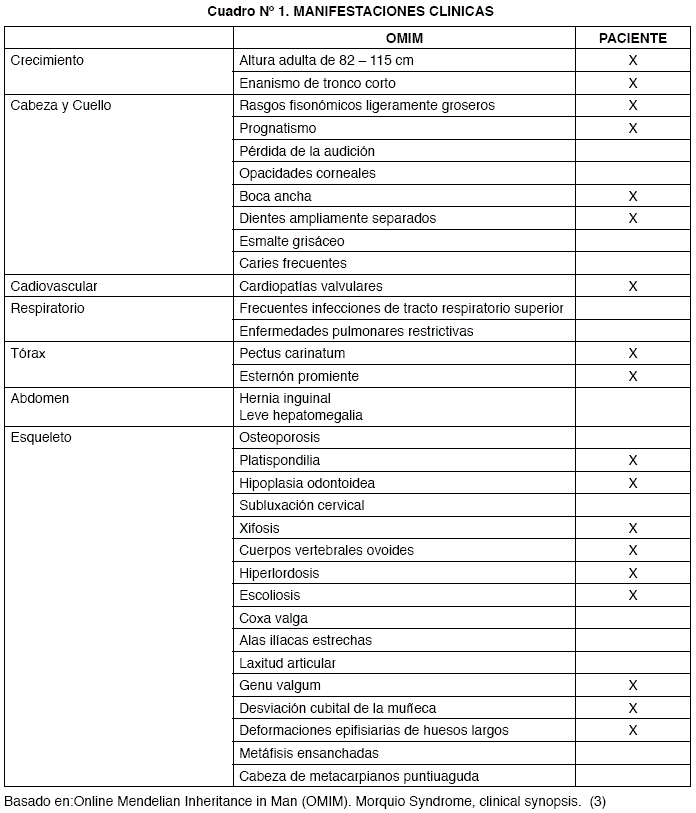
En la Cuadro N°1 se exponen las principales manifestaciones clínicas del síndrome de Morquio. Nos permitimos realizar una contingencia entre lo que reporta la literatura y las manifestaciones de la enfermedad halladas en el paciente.
Escoliosis toracolumbar. Porestas malformaciones se hace imposible tallar adecuadamente al paciente dado que le es imposible pararse completamente erguido.
DISCUSIÓN:
En el medio, para el diagnóstico de un SM nos basamosfundamentalmente en la clínica, dado que actualmente, no poseemos los medios apropiados para detectar los cambios acontecidos a nivel molecular (detección de queratán-sulfato en la orinay actividad enzimáticaen muestra sanguínea). Para el caso presentado se contó con el apoyo del Dr. Néstor Chamoles de la Fundación para el Estudio de Enfermedades Neurometabólicas localizado en Buenos Aires, Argentina; al cual se remitió una muestra sanguínea. Los resultados reportaron negativización ante la prueba para SM tipo B, por lo que inferimos correlacionando esta información con la severidad clínica presente en el caso expuesto, que nos encontrábamos ante un SMtipoA.
Cabe destacar que los cuadros clínicos presentes, fundamentalmente los tipos AyB, son clínicamente indistinguibles entre sí, por lo que resulta casi imposible discernir entre ellos sin la ayuda de laboratorio. Se acepta que los casos de fenotipo más complejo y que reúnen casi la totalidad de los hallazgos clásicamente descritos corresponden al subtipo A, siendo este el más grave (los pacientes no suelen exceder los 25 años de vida). Los casos de tipo C no suelen presentar de manera tan marcada las severas malformaciones descritas en los tipos A y B y estos pacientes pueden tener una supervivencia incluso pasada la sexta década de vida. Por este motivo también se descartó el tipo C en el diagnóstico, pues la abuela falleció a temprana edad. (3)
Las principales características del síndrome las suelen componer las anormalidades esqueléticas: baja talla (particularmente bajo la forma de enanismo de tronco corto), tórax en tonel y pectus carinatum, genu valgo, hiperlaxitud articular y opacidades corneales (11).
El crecimiento está muy comprometido desde los 5 años y la estatura promedio se encuentra entre los 85 y los 100 cm en ausencia de retardo mental. La sordera neurosensorial se inicia aproximadamente al comenzar la segunda década de vida y afecta al 100% de los pacientes después de los 20 años. Una manifestación constante, pero tardía, suelen ser la frecuente aparición de cardiopatía valvular debido al depósito de mucopolisacáridos a nivel de válvulas cardíacas, determinando el desarrollo de insuficiencia de las mismas. Esta última asociada a la severa escoliosis que suele ser motivo de problemas respiratorios constituye la principal causa de mortalidad en estos pacientes (2, 11).
Vale la pena destacar las alteraciones en el desarrollo, encontradas principalmente a nivel de columna cervical y esqueleto del tórax, fundamentalmente por motivo de las severas repercusiones que traen estos defectos durante el desarrollo del individuo. La columna vertebral suele encontrarse severamente afectada, estando los cuerpos vertebrales anómalamente aplanados (platispondilia) debido al acumulo de mucopolisacáridos que debilita su estructura y resistencia a pesos verticales. En ocasiones una o más vértebras se luxan hacia atrás, reduciendo el perímetro del canal medular y condicionando una compresión medular. La columna puede estar curvada hacia un lado (escoliosis), hacia atrás formando una giba (xifosis), o una mezcla de las dos posibilidades (xifoescoliosis). El esternón crece casi normalmente, pero debido a su unión con una columna vertebral escoliótica con severa afección ósea y déficit en su desarrollo tiende a encorvarse hacia adelante adoptando una forma en pico de loro. Por este motivo el tórax adquiere la forma de quilla de barco (pectus carinatum), encontrándose las costillas comprometidas al mantenerse fijas en una posición horizontal, afectando la respiración.
Otro defecto encontrado en este paciente con SM es la hipoplasia o aplasia de la apófisis odontoides del axis. Esta anomalía condiciona una excesiva movilidad del atlas sobre el axis, y finalmente una posible luxación o sub-luxación atlanto-axial. Al suceder este evento, el perímetro del canal medular se ve drásticamente disminuido comprometiendo así a la médula. Debido a la alta localización de este evento el paciente desarrolla una tetraparesia o tetraplejia progresiva que limita severamente su calidad de vida. En severos casos de compresión se puede incluso llegar hasta la parálisis respiratoria debido al compromiso bulbar. El tratamiento de este trastorno suele ser quirúrgico, consistiendo en fijación de las vértebras comprometidas, o en caso de haberse instalado la compresión medular, laminectomía descompresiva entre C1 y C4 (12).
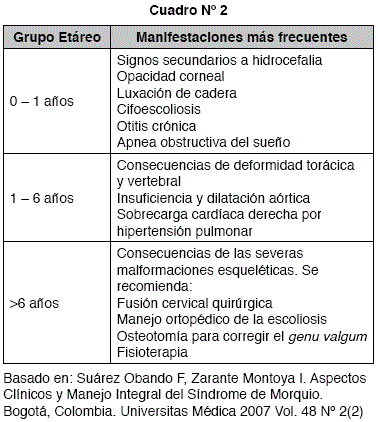
Actualmente no existe cura definitiva para la enfermedad, pero sí tratamiento para los síntomas y complicaciones, a medida que éstas van apareciendo. Se han usado varios métodos experimentales para tratarde administrar la enzima alterada exógenamente, pero sin beneficios significativos a largo plazo (6). El manejo clínico del SM se realiza en base a la aparición paulatina de los trastornos del síndrome, en búsqueda de manifestaciones tempranas que permitan iniciar apoyo precoz para impedir el desarrollo o mitigar el impacto, que generarán las manifestaciones o complicaciones de ellas una vez instauradas. El conocimiento de la historia natural de la enfermedad resulta, entonces, fundamental para poder orientar la búsqueda. En la Cuadro N° 2 se resumen los principales hallazgos según edad del paciente.
Finalmente, si bien esta patología se mantiene hasta la fecha incurable, existen posibilidades de tratamiento y apoyo a estos pacientes, enfocados en disminuir el terrible impacto que esta patología supone sobre su vida. Todo depende de un diagnóstico correcto y precoz. Por este motivo es que instamos a los diferentes servicios de pediatría tener en mente, no solo al SM, sino a todos los trastornos errores innatos del metabolismo en su conjunto. Recordemos que muchos de estos trastornos se manifiestan tempranamente con alteraciones de la postura, los cuales pueden suponer confusión con simples dismorfias no relacionadas con problemas innatos. Por este motivo es fundamental e importante el diagnóstico, no solo para el paciente, sino también para los padres que requieren un adecuado asesoramiento genético que los inste a evitar tener más descendencia.
Lamentablemente en el presente caso, se encontraron en la familia otros afectados como se muestra en el Heredograma que a la fecha se han re captado con ayuda del programa Moto Méndez.
REFERENCIAS
1. Lyons Jones K. Smith's Recognizable Patterns of Human Malformation. 4°Edición. Editorial W.B. Saunders Company. Philadelphia, EEUU. 1988. P.699 [ Links ]
2. Suárez Obando F, Zarante Montoya I. Aspectos Clínicos y Manejo Integral del Síndrome de Morquio. Bogotá, Colombia. Universitas Médica 2007 Vol. 48 N° 2 [ Links ]
3. OMIM #253000. Morquio Syndrome A. Disponible en: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=253000. Consultado 14/04/09. [ Links ]
4. OMIM #252300. Morquio Syndrome C. Disponible en: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=252300. Consultado 14/04/09. [ Links ]
5. Nussbaum R. McInnes R. Williard H. Thompson y Thompson Genética en Medicina. 5ta Edición. Editorial Masson. Barcelona, España. 2004. P. 222-23. [ Links ]
6. Farreras. Rozman. Medicina Interna. 16va Edición. Editorial Elsevier. Barcelona, España. 2009. 1909-17 [ Links ]
7. OMIM #253010. Morquio Syndrome B. Disponible en: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=253010. Consultado 14/04/09. [ Links ]
8. Lyons Jones K. Smith's Recognizable Patterns of Human Malformation. 4° Edición. Editorial W.B. Saunders Company. Philadelphia, EEUU. 1988. P.416-17 [ Links ]
9. Rimoin D. Connor M. Pyeritz R. Korf B. Emery and Rimon 's Principles and Practice of Medical Genetics. 4th Edition. Editorial Churchill Livingstone. China. 2002. P.2672-73 [ Links ]
10. Saul RA. Seaver LH. Sweet KM. Greer JS. Phelan MC. Mills CM. Growth References: Third Trimester to Adulthood. Greenwood Genetic Center. Keys Printing. South Carolina, USA. 1998. Pg 70. [ Links ]
11. Amar Taksande, Krishna Vilhekar. Hurler syndrome or Hurler síndrome. Intelligence required for diagnosting the case. The Indian Journal of Radiology and Imaging. Vol 18, Número 2, Mayo 2008 (Carta al Editor) [ Links ]
12. Imaizumi C. Costa Nova I. de O Joaquim A. Efectos de la fisioterapia en paciente portador de mucopolisacaridosis. Sao Paulo, Brasil. Revista Neurociências Volumen 15, Número 200 [ Links ]

