











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkCuadernos Hospital de Clínicas
versión impresa ISSN 1562-6776
Cuad. - Hosp. Clín. vol.51 no.2 La Paz jul. 2006
ARTICULO ORIGINAL
DNA-UMSAgen, EXTRACCIÓN DE DNA GENÓMICO PARA DIAGNÓSTICO MOLECULAR: MÉTODO RÁPIDO Y ECONÓMICO
Ricardo Amaru *, Hortencia Miguez *, Rosario Peñaloza *, Gina Torres *, Julio Silvestre *, Heriberto Cuevas *.
* Unidad de Biología Celular, Departamento de Ciencias Funcionales, Facultad de Medicina, UMSA,
RESUMEN
INTRODUCCIÓN
El estudio del ácido desoxirribunocleíco (DNA) es imprescindible para la medicina moderna; por ello, se ha diseñado diferentes técnicas para su extracción, cuyos costos son altos y de tiempo prolongado. En este trabajo se describe un método modificado de extracción de DNA (DNA-UMSAgen) basado en la técnica de Miller.
MÉTODOS
Las células mononucleares fueron obtenidas de sangre venosa periférica de un sujeto voluntario, para realizar 40 extracciones de DNA; 20 con el método modificado DNA-UMSAgen y otros 20 con el método clásico. Posteriormente se evaluó la concentración, calidad y utilidad de estos DNA extraídos.
RESULTADOS
La pureza del DNA extraído por el método DNA-UMSAgen es de 1,88 similar al de la técnica clásica 1,91, las concentraciones obtenidas son 20,4 ug/106 cel y 56ug/106 cel respectivamente. La evaluación por electroforesis en agarosa y la amplificación del exon 12 del gen JAK2 por PCR fue satisfactoria.
CONCLUSIÓN
El método DNA-UMSAgen es una alternativa de extracción de DNA genómico, rápido y económico, adecuado para países en vías de desarrollo.
PALABRAS CLAVES
Rev. Cuadernos 2006; 51 (2): 11-15 / DNA, extracción, espectrofotometría, PCR, electroforesis en agarosa
ABSTRACT
INTRODUCTION
DNA is the genetic material of the cell. Actually for its study, laboratory techniques are available that require its extraction free of impurities. This paper describes the DNA-UMSAgen method as an alternative for DNA extraction which is based on the Miller technique.
METHODS
The mononuclear cells were obtained of venous peripheral blood of a voluntary subject, for to realize 40 DNA's extractions; 20 with the modified method DNA-UMSAgen and other 20 with the classic method. Later there was evaluated the concentration, quality and utility of these extracted DNA.
RESULTS
The DNA purity extracted by the DNA-UMSAgen method is 1,88 similar to the classic technique 1,91, the concentration obtained were 20,4 ug/106 cells and 56ug/106 cells respectively. The evaluation by agarose gel electrophoresis and the amplification of the exon 12 of the JAK2 gen by PCR was successful.
CONCLUSION
The DNA-UMSAgen extraction method is a very acceptable fast, easy and inexpensive alternative method for underdeveloped countries for DNA extraction.
KEY WORDS
DNA, extraction, spectrophotometry, PCR, agarose gel electrophoresis.
INTRODUCCIÓN
El ácido desoxirribonucleico (DNA) es el material genético de las células eucarióticas1 cuya manipulación y análisis es imprescindible para el estudio de las bases moleculares en las enfermedades; actualmente diversos métodos de biología molecular permiten extraer DNA desde virus hasta células humanas2,3.
Para la extracción del DNA se requiere aislar proteínas, polisacáridos y lípidos; además, el material utilizado en el proceso de extracción debe estar libre de DNasa asociado a manipulación cuidadosa para evitar su degradación3,4.
A diferencia del método clásico, el método modificado DNA-UMSAgen no utiliza proteínasa K ni fenol cloroformo para la extracción del DNA.
El presente trabajo describe un método modificado de extracción de DNA genómico a partir de la técnica de Miller (5) que es rápido y económico.
MATERIAL Y MÉTODOS
Las células mononucleares fueron separadas por gradiente de concentración en Histopaque 1077 (Sigma - Aldrich, Reino Unido) a partir de 10 ml sangre venosa periférica de un sujeto voluntario, posteriormente las células mononucleares fueron alicuotadas a una concentración de 1 x 106 en tubos Eppendorf y conservadas a
REACTIVOS
- NLB (nuclear lysis buffer)
10 mM Tris-HCl pH 8,2
(Tris-hidroximetilaminometano)
0,4 M Na Cl (Cloruro de sodio)
2 mM Na2 EDTA pH 8,0 (Acido Etilendiamino tetraacético sal disódica)
Disolver - SDS 10% p/v (dodecil sulfato de sodio)
Disolver
Mezclar 300 ml de NLB con 20ml de SDS al 10%, antes de usar. - Na Cl 6M (cloruro de sodio saturado)
Disolver - Acetato de sodio 3M
CH3COONa (acetato de sodio)
Disolver - Solución de lisis para glóbulos rojos 10X
NH4Cl (cloruro de amonio)
KHCO3 (bicarbonato ácido de potasio)
Na2 EDTA pH 8,00 (Acido Etilendiamino tetraacético sal disódica)
Disolver
EXTRACCIÓN DEL DNA GENÓMICO CON EL MÉTODO DNA-UMSAgen.
- Resuspender el sedimento de células (1 x 106) en 900 µl de NLB-SDS.
- Agregar 300 µl de cloruro de sodio
- Mezclar por inversión durante 5 minutos hasta observar una solución homogénea.
- Centrifugar a 4000 rpm durante 5 minutos.
- Recuperar la fase superior en otro tubo Eppendorf.
- Agregar 600 µl de cloroformo - alcohol isoamílico 24:1.
- Mezclar por inversión durante 5 minutos hasta observar una solución homogénea.
- Centrifugar a 4000 rpm durante 5 minutos.
- Recuperar la fase superior en otro tubo Eppendorf.
- Agregar 30 µl de acetato de sodio 3M.
- Agregar 1 ml de alcohol absoluto frío (conservado a
- Mezclar hasta la visualización del DNA precipitado.
- Sacar el DNA precipitado con una aguja hipodérmica G21 y depositarlo en otro tubo Eppendorf.
- Resuspender en 1ml de etanol al 70% (conservado a
- Centrifugar a 12.000 rpm durante 5 minutos.
- Desechar el sobrenadante y dejar secar el DNA a medio ambiente.
- Resuspender en 100 µl de agua tridestilada.
- Dejar una noche a temperatura ambiente para su disolución completa.
- Conservar a
EXTRACCIÓN DEL DNA GENÓMICO CON EL MÉTODO CLÁSICO.
- Resuspender el sedimento de células (1 x 106) en 480 µl de TNE 1x.
- Agregar 100 µl de SDS 10% y 20 µl de proteinasa K (20 mg/ml).
- Incubar en baño maría a
- Centrifugar a 4.000 rpm por 5 minutos.
- Recuperar el sobrenadante en otro tubo Eppendorf.
- Agregar 600 µl de fenol - cloroformo (1:1). Mezclar por 5 minutos hasta su homogenización.
- Centrifugar a 4.000 rpm por 5 minutos.
- Recuperar el sobrenadante en otro tubo Eppendorf.
- Agregar 600 µl de cloroformo/alcohol isoamílico (24:1). Mezclar por 5 minutos hasta su homogenización.
- Centrifugar a 4.000 rpm por 5 minutos.
- Recuperar el sobrenadante en otro tubo Eppendorf.
- Agregar 50 µl de acetato de sodio
- Agregar 1 ml de etanol absoluto frío (
- Centrifugar a 12.000 rpm por 5 minutos.
- Desechar el sobrenadante. Agregar 1ml de etanol al 70% conservado a
- Centrifugar a 12.000 rpm por 5 minutos.
- Desechar el sobrenadante.
- Secar el DNA a medio ambiente.
- Agregar 100 µl de agua tridestilada y dejar por una noche a temperatura ambiente para su disolución completa.
- Conservar a
ANÁLISIS CUANTITATIVO DEL DNA.
La concentración de DNA se determinó por espectrofotometría UV a 260 nm en una solución diluida de DNA 1/50. El cálculo se realizó con la siguiente fórmula:
![]()
ANÁLISIS CUALITATIVO DEL DNA
La calidad del DNA se evaluó mediante la separación electroforética en gel de agarosa al 0.8% con bromuro de etidio, migrado a 80 V por 60 minutos. En la migración se utilizó el marcador de pares de bases: Marker II. Se consideró DNA de alta calidad, a la presencia de bandas iguales o superiores a 23.000 bp.
ANÁLISIS DE PUREZA DE DNA
La pureza del DNA se calculó mediante la relación entre la absorbancia a 260 nm y 280 nm; considerando DNA de alta pureza a valores igual a 1.8 ± 0.1.
AMPLIFICACIÓN DE DNA
El DNA genómico se amplificó mediante la técnica del PCR (reacción en cadena de polimerasa) utilizando primers del exon 12 del gen Jak2 de acuerdo a protocolo establecido6. Los productos amplificados fueron separados por electroforesis en gel de agarosa al 2% con bromuro de etidio.
RESULTADOS
Se realizaron 40 extracciones de DNA genómico a partir de 1 x 106 células mononucleares, 20 por el método DNA-UMSAgen y 20 por el método clásico.
Las concentraciones de DNA fueron 20.4 µg y 56 µg por 106 células mononucleares por el método DNA-UMSAgen y Clásico respectivamente (cuadro 1 y 2). La pureza del DNA extraído fue 1.88 para el método DNA-UMSAgen y 1.91 por el método clásico (cuadro 1 y 2).
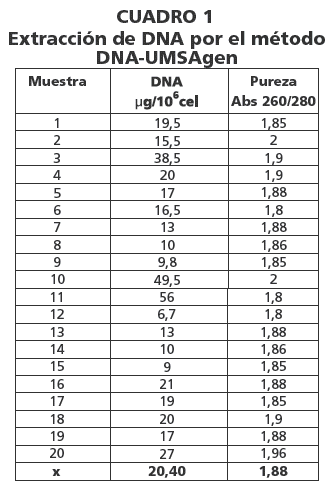
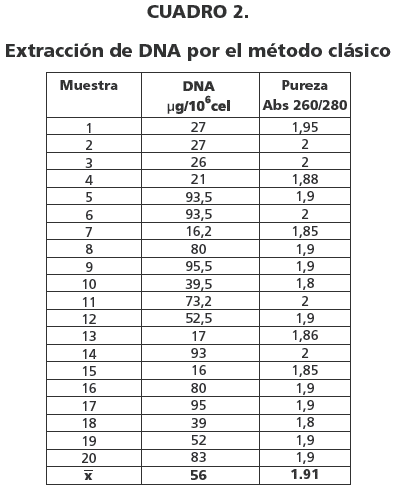
Los resultados de la concentración del DNA extraído por ambos métodos son estadísticamente diferentes y en referencia a la pureza y calidad ambos métodos son adecuados (cuadro 3, figura 1).

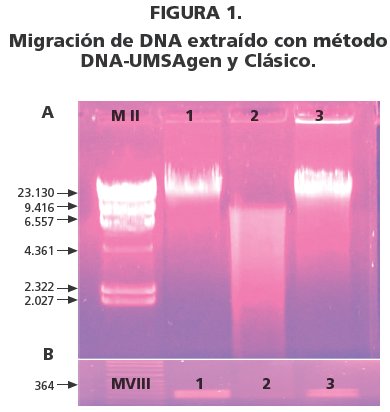
- Migración electroforética de 3 µg de DNA en agarosa 0.8% con bromuro de etidio. MII, Marker II; columna 1, método DNA-UMSAgen; columna 2, DNA degradado; columna 3, método clásico.
- Migración electroforética de Amplificación del exon 12 del gen JAK2 por PCR en agarosa 2% con bromuro de etidio. MVIII Marker VIII; columna 1, método DNA-UMSAgen; columna 2, DNA degradado; columna 3, método clásico.
El estudio de costos para el método DNA-UMSAgen es de un dólar y para el método clásico es de dos dólares.
El tiempo de extracción utilizado por el método DNA-UMSAgen es de 2 horas, mientras para el método clásico es de 24 horas.
DISCUSIÓN
El DNA genómico es utilizado en laboratorios de Biología Molecular para determinar la etiología y las características genéticas de diferentes patologías.
Desde la primera extracción del DNA hasta la fecha se han diseñado diversos métodos de extracción; en los últimos años se hicieron populares los Kits patentados que simplifican la extracción, pero disminuyen la calidad del DNA con costos cada vez mayores.
En el método DNA-UMSAgen no se utiliza la proteinasa K, (enzima de alto costo); en su lugar se utiliza el NLB (del anglosajón: "nuclear lysis buffer") que reduce el tiempo del procedimiento de 12 horas (método clásico) a 5 minutos. Además no se utiliza el fenol, una sustancia catalogada como muy tóxica para el manipulador y para el medio ambiente. Al recuperar el DNA precipitado con una aguja hipodérmica, nos permite obtener DNA de alta calidad (de mayor número de pares de bases) dejando DNA de menor número de pares de bases en la solución; probablemente por ello la concentración del DNA sea inferior en relación con el método clásico.
El método DNA-UMSAgen simplifica los pasos, ahorra tiempo y dinero, obteniendo DNA de alta calidad tal como lo demuestran los estudios de espectrofotometría y electroforesis en gel de agarosa. Los costos de extracción se reducen en aproximadamente 50% en relación con el método clásico y más de 200% en relación con otros métodos patentados.
La cantidad de extracción de DNA con DNA-UMSAgen es menor en relación con el método clásico, pero suficiente para realizar técnicas como Southern blot, PCR, etc. La pureza es ligeramente superior con DNA-UMSAgen aunque no estadísticamente significativo.
El método DNA-UMSAgen con las ventajas ya descritas, requiere ser validado para su utilización a nivel internacional.
REFERENCIAS.
1. Watson JD, Crick FHC. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953; 171: 737.
2. Jones SW, Dobson ME, Francesconi SC, Schoske R, Crawford R. DNA Assays for Detection, Identification, and Individualization of Select Agent Microorganisms Croat Med J 2005; 46: 522.
3. Maniatis T, Fritsch EF, Sambrock J. Molecular cloning: A laboratory Manual. Ed. 1982. Cold Spring Harbor. New York. [ Links ]
4. Luque J, Herraez A. Texto ilustrado de Biología Molecular e Ingeniería Genética. Ed. 2001. Harcourt. Madrid.
5. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res 1988; 16: 1215.
6. Baxter EJ, Scott lm, Campbell PJ, et al. Adcquired mutation of the tyrosine kinase JAK-
 Todo el contenido de esta revista, excepto dónde está identificado, está bajo una Licencia Creative Commons
Todo el contenido de esta revista, excepto dónde está identificado, está bajo una Licencia Creative Commons
Av. Saavedra Nº 2246, Miraflores Teléfono IP 2612309
La Paz - Bolivia

revista.cuadernos@umsalud.edu.bo