











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkCuadernos Hospital de Clínicas
versión impresa ISSN 1562-6776
Cuad. - Hosp. Clín. vol.51 no.1 La Paz 2006
ARTICULO ORIGINAL
Manifestaciones clínicas más frecuentes en pacientes con sindrome de Turner del Instituto de Genética,
Univ. Castillo Zalles Claudia * Univ. Ramos Quisbert Geraldine * Univ. Vila Chipana Wilder *, Dr. Javier Mercado Gordillo** Dra. Ximena Aguilar Mercado ***
*Auxiliar investigador del Instituto de Genética
**Residente primer año de Medicina Interna - Hospital de clínicas
***Docente Investigador Titular del Instituto de Genética - Master en Ciencias Biológicas Biomédicas mención Genética Médica
RESUMEN
Objetivo
Identificar los signos clínicos más frecuentes en pacientes con Síndrome de Turner.
Diseño
Corte transversal.
Lugar
Instituto de Genética;
Población
36 pacientes con diagnóstico citogenético.
Métodos
Recolección de datos clínicos de pacientes con Síndrome de Turner períodos 1990-2004. Se excluyeron pacientes que presentaban similares fenotipos y con cariotipo no compatible.
Resultados
Manifestaciones clínicas más frecuentes: baja talla proporcionada 77.8%, disgenesia gonadal 61.1%, pterigium colli 27.8%, displasia de pabellones auriculares 33.3%. La edad de diagnóstico corresponde: < 5años 11.1%, entre
Conclusión
Clínicamente el Síndrome de Turner es variable, y es diagnosticado más frecuentemente durante la adolescencia, etapa en la que se perdieron oportunidades para un adecuado tratamiento que coadyuve a prevenir complicaciones.
El fenotipo de esta cromosomopatía actualmente a sido relacionado con mutaciones de genes como RPS4X y SOS .
Palabras clave
Rev. Cuadernos 2006;51(1)27-32, Síndrome de Turner, fenotipo, citogenética, disgenesia gonadal, baja talla.
ABSTRACT
Objective
Identify the most frequents clinical Turner syndrome patients features.
Design
Cross section.
Place
Genetic Institute,
Participants
36 patients with Turner Syndrome.
Methods
Clinical features data were collected from Genetic Institute records. Patients with similar clinical features with out cariotyping diagnosis where not included.
Results
We find small stature 77.8%, gonads dysgenesis 61.1%, pterigium colli 27.8%, anomalous auricles 33.3%. Age of diagnosis was less than 5 years 11.1%, between to
Conclusions
The clinical features of Turner Syndrome are variable, the diagnosis it's most frequency during puberty, age where could it be late to prevent consequences. The phenotype of Turner Syndrome has been related to RPS4X and SOS genes.
Key words
Turner Syndrome, cytogenetic analysis, features, gonads dysgenesis, small stature.
INTRODUCCIÓN
El presente trabajo tiene el objetivo de informar sobre las manifestaciones más frecuentes del Síndrome de Turner (ST) en nuestro medio, durante el periodo 1990-2004, enfatizando en la importancia del diagnóstico precoz de este síndrome, y la repercusión positiva en el seguimiento y prevención de complicaciones, piezas claves en la mejora de la calidad de vida de las afectadas.
La diferenciación sexual humana, se debe a la presencia e interacción de los genes presentes en los cromosomas X, Y y en otros autosomas, que resultan en un genotipo de 46 XX para la mujer y de 46 XY para el varón. Existen casos en los que la carga cromosómica se encuentra alterada en forma cuantitativa pudiendo observarse con frecuencia tanto las trisomías como las monosomías cromosómicas; precisamente dentro de este último grupo se sitúa el ST, que tiene una incidencia de 1 en 2000 recién nacidos vivos (r.n.v).(1)
El ST se manifiesta con cariotipo característico (45 XO) en un 45%; como mosaico
El ST fue descrito por primera vez por Henry Turner en 1938 como un conjunto de caracteres fenotípicos entre los que podemos citar: Talla baja proporcionada, disgenesia gonadal y un patrón de malformaciones principales y secundarias. Las más frecuentes son renales, cardiacas, óseas, otorrinolaringológicas y otras.
Las características clínicas varían ampliamente según la edad y la anomalía citogenética que presentan las pacientes con ST. Los "estigmas" turnerianos son característicos de las pacientes con monosomía Xy con isocromosoma del Xq; los pacientes con delección del Xq a menudo solo presentan disgenesia gonadal (2). Resaltar esta amplia variabilidad genetica es importante para entender la amplia variedad fenotípica encontrada en la practica clínica diaria.
El fenotipo clásico del ST se relaciona con ausencia total o parcial de la porción proximal del brazo corto de los cromosomas X o Y. Siendo en este caso el fenotipo de la enfermedad consecuencia de la pérdida de genes homólogos, localizados en estas regiones cromosómicas, mismos que deben encontrarse en doble dosis para permitir un desarrollo normal. La pérdida de alguno de estos genes se ha denominado haploinsuficiencia. En la actualidad se realizan estudios moleculares encaminados a identificar genes implicados en el desarrollo de la enfermedad. A continuación se mencionan brevemente algunos de estos genes: El ZFX y sus homólogo ZFY, localizados en los cromosomas X y Y (3). Ambos actúan durante la diferenciación sexual. El RPS4X y SOS que tienen un homólogo funcional en el cromosoma Y y escapan al proceso de inactivación del X.(7,8,9).
Con relación a la talla baja Rao y cols. identificaron el gen SHOX en el cromosoma (Xp) en la región PAR-1 (Pseudo Autosomic Region - que escapa a la inactivación), que participa activamente en el desarrollo y crecimiento óseo predominante en miembros superiores. (6). Datos actuales encontraron que SHOX en haploinsuficiencia está relacionado con talla baja, 4° metacarpiano corto, cúbito valgo y discondrosteosis de Lery-Weill. La expresión del SHOX es más evidente en la porción media de los miembros (mesomelia) y en el primer y segundo arco faríngeo (7).
Clinicamente las pacientes con isocromosoma Xp y con delección del brazo largo del cromosoma X no presenta los "estigmas"somáticos del ST, pero sí disgenesia ovárica. Por otra parte, en el mosaicismo con línea celular normal el cuadro clínico es más atenuado. Se debe tomar en cuenta que en mosaicismos que implican a parte o a todo el cromosoma Y, existe riesgo de desarrollar gonadoblastoma (2).
METODO
El presente es un estudio de corte transversal, realizado en el Instituto de Genética (IG) de
Para este estudio se revisaron un total de 1577 historias clínicas del IG. Se incluyeron en el estudio 36 historias que correspondían a pacientes con el diagnóstico citogenético y clínica acorde con ST. Se excluyeron pacientes de ambos sexos con similares características clínicas, sin alteraciones cromosómicas.
RESULTADOS
Encontramos un total de 36 pacientes con diagnóstico citogenético de ST, que representa al 2.35% del total de pacientes que fueron atendidos durante el período 1990 al 2004 en nuestra institución.
Entre las manifestaciones clínicas se tomaron en cuenta los 35 signos clínicos reportados como los más frecuentes de acuerdo a bibliografía consultada, se compararon los porcentajes encontrados con los referidos en la literatura, correlacionados con el número de pacientes del estudio portadores de dicho signo en la tercera columna. (ver Cuadro 1).
Obtuvimos mayor frecuencia diagnostica de ST
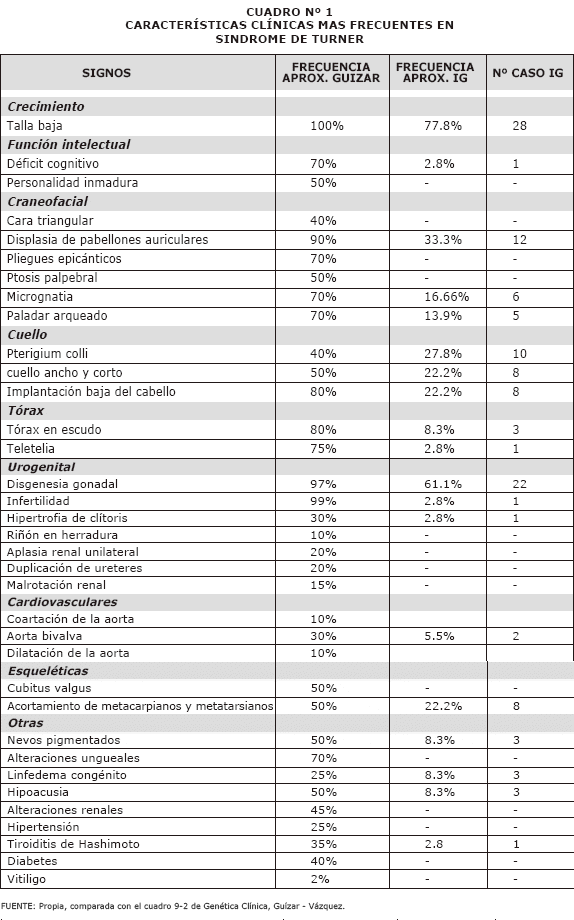
En cuanto a los datos obtenidos sobre la edad en la que se realizó el diagnostico esta detallada en el Gráfico N° 1. entre
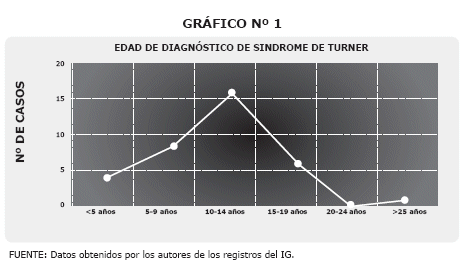
El estudio citogenético se realizó al 100% de los pacientes estudiados (36 casos), con mayor porcentaje de monosomías simples 72%, seguido de alteraciones en mosaico 28%, no se encontró ninguna alteración por delección. Gráfico 2
DISCUSIÓN
Los hallazgos de este estudio ratifican la amplia variabilidad clínica que puede traducirse en un diagnóstico tardío y/o en una no adecuada prevención de las complicaciones que presentan las pacientes con ST.
En el análisis el signo físicos más frecuente fue talla baja 77.8%, que no concuerda con el 100% reportado por Guizar, nosotros consideramos que la talla baja también debe ser considerada dentro de la variabilidad clínica del ST sustentada por nuestros hallazgos. En cuanto a la disgenesia gonaldal 61.1% e infertilidad 2.8%, encontrada en este estudio consideramos que el seguimiento y los estudios complementarios que se realiza de estas pacientes incidirán en el incremento de este porcentaje.
Otros de los signos con mayor frecuencia encontrados fueron displasia de pabellones auriculares 33.3% y Pterigium Colli 27.8%.
En cuanto a la edad en que se realiza el diagnóstico en nuestros datos el signo de sospecha diagnóstica más frecuente encontrado entre los 5-9 años es la talla baja proporcionada y el linfedema en recién nacidas que correspondió al 11%.
Para concluir consideramos que la variabilidad de signos clínicos en el ST, en la practica clínica cotidiana reviste gran importancia debido a todos los factores biopsicosociales que reviste esta entidad genética que se manifiesta al nacer, pudiendo causar discapacidad para toda la vida(4). Concretamente nos referimos a las manifestaciones cardiacas y la disgenesia ovárica; ambas pueden ser seguidas adecuadamente en el medio por los especialistas respectivos si se utilizan las herramientas diagnósticas a nuestro alcance, apoyados con el diagnóstico citogenético considerado como gold standart (4).
Consideramos como diagnóstico tardío el realizado entre los 10 y 14 años, pues se perdieron a esta edad las oportunidades de tratamiento y seguimiento de las cardiopatías, probabilidad de terapia hormonal para mejorar la talla final, causa de problemas de aceptación del esquema corporal, pérdida de oportunidades en la intervención para estimular una adecuada diferenciación sexual secundaria a través del manejo hormonal sustitutivo en forma cíclica por falla ovárica.
La oportunidad de asesoramiento genético es importante para que la paciente y la familia entiendan las limitaciones reproductivas, problemas en el desarrollo maxilofacial, búsqueda de malformaciones a nivel genitourinario. Por otro lado se debe considerar junto al equipo médico las medidas encaminadas a la prevención de la osteoporosis cuando las pacientes alcancen la edad adulta.
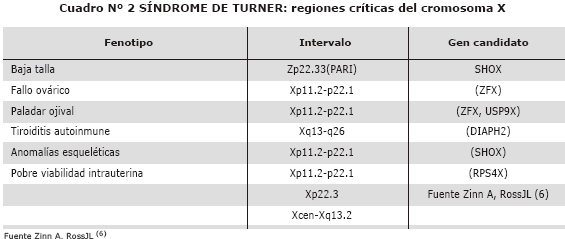
Recomendamos tomar en cuenta la relación de talla baja con el gen SHOX, si la anomalía cromosómica reside exclusivamente en el brazo largo la talla será normal y no expresará el fenotipo turneriano. Respecto a la disgenesia ovárica existen genes candidatos en el brazo corto y en el brazo largo del cromosoma X (Ver cuadro 2).
Se resumen a continuación los aspectos a tomar en cuenta frente a una paciente con ST: Valoración periódica a diferentes edades y manejo multidisciplinario, permanente al momento del diagnóstico. Pedir una evaluación por cardiología y nefrología, evaluar durante el seguimiento la posibilidad de otitis media y otitis serosa que son causas frecuentes de hipoacusia. El endocrinólogo será el encargado de evaluar la falla gonadal, tomando en cuenta el apoyo hormonal para el desarrollo de los caracteres sexuales secundarios. Considerar realizar cirugía plástica del cuello en caso de afectación de la estética corporal.
El Asesoramiento Genético, estará dirigido a guiar a la paciente y familiares en cuanto a la esfera reproductiva, talla, esquema corporal y prevención de enfermedades concomitantes, además de estimular la formación de grupos de autoayuda.
REFERENCIAS
1. D. Smith: "Smith's Recognizable Patterns of Human Malformation"; 5th Edition; edit. W. B. Saunders; 1997; pg. 74 - 79. [ Links ]
2. J. Solari: "Genetica Humana. Fundamenteos y aplicaciones en Medicina"; 3ra edición: edit. Panamericana; 2004; Pág.392-394.
3. J. Guizar: "Genetica Clínica"; 3ra Edición; edit. Manual Moderno; 2001 [ Links ]
4. Informe de un Grupo Cientifico de
5. Zinn A, Ross JL: "Critical regions for Turner syndrome phenotypes on the X chromosome"; eds. Optimizing health care for Turner patients in the 21st century.
6. Kosho T, Muroya K y cols.: "Skeletal features and growth patterns in 14 patients with haploinsufficiency of SHOX:implications for the development of Turner Syndrome"; J Clin Endocrinol Metab; 1999; 84:4613-4621
7. Dr. Y. ALBISU Jefe de Servicio de Pediatría Hospital Nª Sra. de Aranzazu Donosita; "SINDROME DE TURNER. DEL GENOTIPO AL FENOTIPO"; actualizado el 27 de enero del 2006; disponible en http://www.svnp.es/Documen/turner.htm.
8. Mac Laughlin D, Donahoe P. Sex Determination and Differenctiation.
9. Bowels J, Koopman P. New clues to the puzzle of mammalian sex determination. http://genomebiology.com/2001/2/9/reviews/1025.3c

