










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO Compartir

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
versión On-line ISSN 1024-0675
Rev. bol. ped. vol.55 no.1 La Paz 2016
CASO CLÍNICO
Agenesia de glándula tiroides: Presentación de un caso clínico
Agennesis of gland thyroid: a case report and review
Drs.: Jennifer Wendy Davila Yamal*, Jaime Rada Cuentas**
* Médico Residente de Tercer Año de Pediatría del Hospital Municipal Boliviano Holandés
** Pediatra-Infectólogo y Epidemiólogo. Coordinador de la Residencia Médica de Pediatría del Hospital Municipal Boliviano Holandés
Artículo aceptado para su publicación el 15/03/2016
Resumen
El hipotiroidismo congénito es la enfermedad endocrinológica más frecuente en pediatría y la primera causa de retardo mental prevenible. El diagnóstico precoz se establece mediante el cribado neonatal de los niveles de hormona tiro-estimulante (TSH), porque solamente un pequeño porcentaje de niños (as) presenta sintomatología clínica en este grupo etario.
Se describe el caso clínico de una niña atendida en el Hospital Boliviano Holandés a quien se le diagnosticó hipotiroidismo congénito secundario a una agenesia tiroidea a los 31 días de vida y se empezó tratamiento de sustitución con levotiroxina sódica, para evitar las secuelas neurológicas posteriores. Se presenta el caso para resaltar la importancia del diagnóstico precoz y la utilidad de implementar el cribado neonatal en forma universal.
Palabras clave:
Rev Soc Bol Ped 2016; 55 (1): 15-22: hipotiroidismo congénito, agenesia tiroidea, levotiroxina.
Abstract
The congenital hypothyroidism is the most frequent endocrinological disease in pediatrics and in turn it is the first reason of mental delay prevenible. The precocious diagnosis establishes by means of the screening neonatal of the levels of hormone shot - stimulant (TSH), because only a small percentage of children presents clinical symptomatology in this group etario. There is described the clinical case of a girl attended in the Bolivian Dutch Hospital who was diagnosed congenital to 31 days of life and treatment of substitution was begun with levothyroxina sodium, to avoid the neurological later(posterior) sequels. One presents the case to highlight the importance of the precocious diagnosis and the of implementing the screening neonatal in universal form.
Key words:
Rev Soc Bol Ped 2016; 55 (1): 15-22: congenital hypothyroidism, thyroid agenesis, levothyroxine.
Introducción
El hipotiroidismo congénito (HC) se define como la deficiencia de hormona tiroidea al nacimiento.1 En general, este desorden es permanente y es el resultado de una alteración en el desarrollo de la glándula tiroides (disgenesia o agenesia)1-2 o un desorden en la biosíntesis de la hormona tiroidea (dishormono-génesis).2 Ambas alteraciones producen hipotiroidismo primario.2 En el 85% el HC se produce por disgenesia tiroidea que puede manifestarse de diferentes formas, dos tercios son causados por ectopia de la glándula tiroidea que con frecuencia se encuentra en la base de la lengua, mientras otros pacientes presentan agenesia (atirosis) o hipoplasia de la glándula tiroides.3 La mayoría de estos casos son esporádicos y sólo el 2% de ellos tienen una reconocida mutación genética.3-4 Los pacientes con atirosis con mayor frecuencia presentan malformaciones congénitas.3 El HC con localización normal de la glándula tiroidea, forma parte de grupo de condiciones heterogéneas y habitualmente se presenta en el 30 a 40% de los casos de hipotiroidismo congénito.3
El hipotiroidismo secundario o central al nacimiento se origina por una deficiencia de la hormona estimulante tiroidea (TSH). El hipotiroidismo periférico es el resultado de los defectos de transporte, metabolismo o acción de la hormona tiroidea.2
Rara vez la alteración de la función tiroidea es transitoria y se presenta por el pasaje transplacentario de medicación materna, anticuerpos bloqueadores maternos, deficiencia o exceso de yodo.2 Debe tenerse en mente que la causa principal de HC no se logra determinar en la mayoría de los casos. Así, la etiología de la mayoría de los casos de disgenesia tiroidea permanece desconocida, aunque parece tener un origen genético.5
Las hormonas tiroideas son esenciales para el crecimiento y desarrollo neurológico de los niños. La glándula tiroides empieza a formarse a las 7 semanas y la hormona tiroidea (tiroxina T4) se produce a las 12 semanas de gestación.6 La disfunción tiroidea en el recién nacido, lactante o niño tiene un impacto significativo sobre el desarrollo,6 siendo el objetivo principal del tratamiento temprano, para asegurar el normal crecimiento y evitar un retardo del desarrollo.6 El hipotiroidismo congénito constituye, la mayor causa prevenible de una potencial discapacidad intelectual.6 La T4 es importante en la mielinización del sistema nervioso central durante los primeros 3 años de vida.6
Las concentraciones de TH son bajas en el feto durante la primera mitad del embarazo.2 Durante este período, es totalmente dependiente de la TH materna; el suplemento hormonal es controlado por la placenta y el estado tiroideo de la madre.2 El eje fetal hipotálamo-pituitaria-tiroides inicia su función en la mitad de la gestación y es maduro en el recién nacido de término al nacimiento.2 Pese a ser tan importante la TH sobre múltiples órganos, especialmente para el cerebro, la mayoría de los lactantes con HC se encuentran normales al nacimiento.2 El feto hipotiroideo parece estar protegido -al menos parcialmente- por la transferencia de TH materna.2 En el período neonatal temprano, las manifestaciones clínicas del HC son sutiles y usualmente indetectables.7 Las complicaciones como la deficiente capacidad intelectual, moderado o severo retardo psicomotor y trastornos en el neurodesarrollo (pérdida de la audición, encefalopatía, epilepsia o desórdenes psiquiátricos), disminución de la velocidad del crecimiento, retardo en la maduración ósea y compromiso de la talla,3 se presentan posteriormente. Estas manifestaciones irreversibles pueden evitarse si el hipotiroidismo es identificado a tiempo y es tratado de preferencia antes de las dos semanas de vida.3,7
Caso Clínico
Lactante femenina de 1 mes de edad. Nacida de una madre de 27 años los antecedentes prenatales informan: control regular en 10 oportunidades, no refiere exposición a sustancias tóxicas, un embarazo prolongado, por ello el parto fue distócico por cesárea. La niña nació con peso de 3.900 g y 51 cm de talla en un centro hospitalario privado. La recién nacida es producto del primer embarazo, se desconoce historia postnatal. Sin embargo, la madre refiere respiración y llanto inmediatos al nacimiento. Cursa con ictericia las 2 primeras semanas de vida, recibiendo tratamiento con fenobarbital en gotas. No se realiza tamizaje de hormonas tiroideas neonatales. Se alimenta con seno materno exclusivo, el desarrollo psicomotriz es adecuado para su edad.
Acude al servicio de pediatría del Hospital Municipal Boliviano Holandés por un cuadro clínico de una semana de evolución, caracterizado por accesos de tos cianozante que interrumpe el sueño y estreñimiento. Se corrobora a su ingreso las características de la tos. Asimismo, la lactante presenta llanto débil y ronco, sin alteraciones en la succión y deglución. Al examen físico, la niña se encontraba en buen estado general, afebril, piel y mucosas hidratadas normo-coloreadas, facies abotagada, llanto débil ronco, cabello abundante, seco, frágil, de implantación baja. Fontanela anterior aproximadamente de 1.5 cm de ancho y la posterior de 2 cm de ancho.
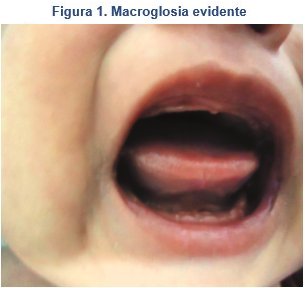
Un perímetro cefálico de 38 cm. En la cavidad oral presenta macroglosia (Figura 1); tórax simétrico, área cardiaca con ruidos cardiacos rítmicos normo fonéticos sin soplos, campos pulmonares bien ventilados con murmullo vesicular conservado.
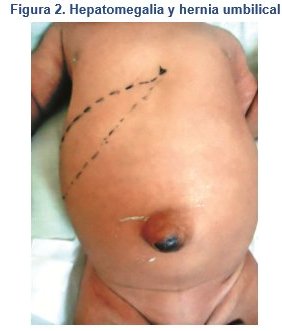
Abdomen blando, depresible, globoso con RHA positivos y normoactivos, se palpa hígado a 2-3-4 cm por debajo de reborde costal, se evidencia hernia umbilical de 1.5 cm de diámetro, sin compromiso intestinal (Figura 2). Extremidades con tono y trofismo conservados y reflejos presentes normales.
Exámenes de laboratorio y gabinete: en el hemograma se informa, una hemoglobina 10.6 g/dL, hematocrito 33%, VES 13 mm/h, glóbulos blancos 8.500/(µL, segmentados 33%, linfocitos 64%, Monocitos 2% y eosinófilos 1%. La determinación de hormonas tiroideas séricas informó: TSH: 36 µUI/L, T3: 1 µg/dL, T4 total 2.9 µg/dL. Bilirrubinas: BT 6.6 mg/dL; BI 4.3 mg/dL y BD 2.3 mg/dL. El cultivo y la PCR para Bordetella pertussis fueron negativas. En la radiografía de tórax no se observaron signos radiológicos de bronquiolitis. La gammagrafía tiroidea de proyección anterior de cuello, post administración de 1.03 mCi de 99mTcO4, no muestra captación de tejido tiroideo e informa agenesia de glándula tiroides. La edad ósea valorada por radiografía de huesos largos no muestra áreas de osificación para la edad. Al quinto día de internación se administra levotiroxina (LT4) a dosis pediátrica, 10 µg /Kg /día, ferrasol 10 mg VO cada 24 horas y glicerina vía rectal con catarsis positiva. El electrocardiograma no muestra ninguna alteración.
Sus diagnósticos de ingreso fueron: bronquiolitis, síndrome coqueluchoide, y anemia clínica. Los de egreso: Resfrío común, anemia ferropriva e hipotiroidismo primario congénito por agenesia de glándula tiroides. Se descartó bronquiolitis y síndrome coqueluchoide.
Las pruebas de control de función tiroidea luego de iniciado el tratamiento se encontraron dentro de parámetros adecuados para la edad.
Discusión
La incidencia de HC es de 1 en 2000 o 4000 nacidos vivos en ciudades donde se implementaron programas de detección temprana neonatal.1 Varía de acuerdo a la localización geográfica y es una de las enfermedades frecuentes, cuyo tratamiento oportuno, previene el retardo mental.1
El HC es más frecuente en niñas con una relación de 2:1 respecto a los niños.1 Sin embargo, de acuerdo al informe de Quebec las mujeres -en general- presentan ectopia tiroidea y con menor frecuencia agenesia de la glándula.1 Las madres mayores de 39 años tienen mayor incidencia de hijos (as) con HC (1:1328) respecto a las madres jóvenes menores de 20 años (1:1703).1 La incidencia es mayor en premaduros versus lactantes de término.8 No está claro si el HC en el premaduro es transicional o permanente.1
Aunque los infantes durante su etapa gestacional no pueden producir hormona tiroidea, más del 95% de los recién nacidos con HC no presentan al nacimiento, manifestaciones clínicas evidentes de hipotiroidismo porque la T4 materna cruza la placenta y les permite mantener concentraciones normales del 25% al 50% que le ofrecen una protección temporal.3,9 Por ello, si el diagnóstico clínico se retrasa hasta los 3 meses de vida o más tiempo, la deficiencia de hormona tiroidea produce efectos irreversibles en el desarrollo del cerebro.9 La talla y peso se encuentran en rango normal, pero el perímetro cefálico puede estar incrementado.6 Una fontanela posterior abierta en un neonato de término podría ser una señal de HC.6 Un reducido porcentaje de infantes puede presentar signos y síntomas que incluyen: somnolencia, letargia, hipotonía, llanto disfónico, problemas de alimentación (no se despierta para lactar, pobre o lenta alimentación) constipación, macroglosia, hernia umbilical, piel seca, hipotermia, extremidades frías e ictericia prolongada con o sin facies grosera/edematosa.6,10 La persistencia de la fontanela posterior, fontanela anterior grande y una sutura sagital ancha reflejan un retardo en la maduración ósea. La ausencia de una o ambas epífisis de la rodilla se relaciona con la concentración de T4 en el momento del diagnóstico, la evolución del coeficiente intelectual e hipotiroidismo intrauterino.10 Se recomienda realizar estudios imagenológicos como la ultrasonografía y cintigrafía tiroidea con yodo-123 y tecnecio-99m radiactivos porque nos permiten determinar la etiología específica.6,10 Con la ecografía determinamos la presencia, localización normal, forma y tamaño de la glándula.11 La cintigrafía tiroidea con yodo-123 y tecnecio-99m, nos ayuda a delinear la etiología de la disfunción tiroidea en el hipotiroidismo primario (agenesia y ectopia) o un defecto completo de dishormonogénesis con bocio palpable, aunque este último hallazgo es infrecuente.6,10-11 Sin embargo, el bocio típicamente se desarrolla en etapa tardía si el paciente no es tratado.6 Por ello, se aconseja realizar un minucioso examen físico a los neonatos y lactantes con HC, para identificar cualquier malformación/síndromes dismórficos o alteraciones del neurodesarrollo.3
Si bien, el 85% de los pacientes de HC primario permanente es debido a una anormal formación de la glándula (disgenesia tiroidea).7 El resto 10% a 15% de casos se debe a una alteración de la biosíntesis de hormona tiroidea (dishormonogénesis tiroidea).7
El HC puede ser clasificado en transitorio y permanente. En el HC transitorio, el estado eutiroideo retorna después de pocas semanas a meses, mientras en el permanente, la deficiencia es de por vida.7
El HC puede ser clasificado como primario, cuando la deficiencia de la hormona tiroidea se debe a disgenesia o dishormonogenesis o HC secundario (llamado también central o HC hipopituitario) si la deficiencia de TSH se debe a un hipopituitarismo congénito.7
Cerca de dos tercios de los casos de disgenesia tiroidea se deben a tejido tiroideo ectópico y el resto es debido a una aplasia o hipoplasia.7
El diagnóstico de este padecimiento se realiza en el recién nacido (RN), interpretando los valores de TSH en una prueba de sangre capilar obtenida del talón a las 48 a 72 horas de vida.12 En los RN premaduros, gemelos o en estado crítico, se aconseja repetir la prueba a las 2 semanas para descartar elevaciones tardías de TSH.12
La determinación de TSH no permite detectar los casos de HC central, la hipotiroxinemia frecuente del RN de bajo peso, ni a los pacientes con elevación tardía de TSH. Por ello, en algunos países, el programa para la detección de HC, evalúa en un inicio T4 que permite -además- la detección de estos padecimientos.12 Cuando el valor de T4 es menor al punto de corte definido para la edad gestacional se repite la prueba capilar.10 Solo se realiza la determinación de TSH en aquellos recién nacidos que presentan un valor de T4 menor al 10% del correspondiente al día de su evaluación. Cuando la concentración de TSH del infante se encuentra normal o elevada, se realiza una nueva prueba de laboratorio confirmatoria. Si la prueba de TSH inicial es normal y en la segunda prueba se encuentra elevada, debe pensarse en un probable HC primario transitorio (en la mayoría de los casos) o un probable HC permanente, aconsejándose iniciar en ambos casos tratamiento lo más pronto posible. En aquellos neonatos con niveles de T4 capilares bajos persistentes, deben determinarse los niveles séricos de T4 y TSH.1,13
El estudio de confirmación diagnóstica incluye: a) anamnesis familiar (enfermedades tiroideas y auto-inmunitarias); b) Niveles séricos de T4 libre, TSH, TBG, anticuerpos antitiroideos; c) gammagrafía tiroidea y ecografía; e) radiografía de rodillas y cálculo de la superficie de la epífisis distal del fémur (mm2) como indicador de la antigüedad del HC.12
Todos los niños con HC a los 3 años requieren una reevaluación diagnóstica para descartar los HC transitorios, excepto en las disgenesias tiroideas confirmadas. No se debe olvidar que antes de realizar una prueba de laboratorio que permite valorar la función tiroidea, debe suspenderse la medicación durante 4 semanas.12 En la figura 3 se esquematiza el algoritmo diagnóstico del cribado neonatal del HC primario.12
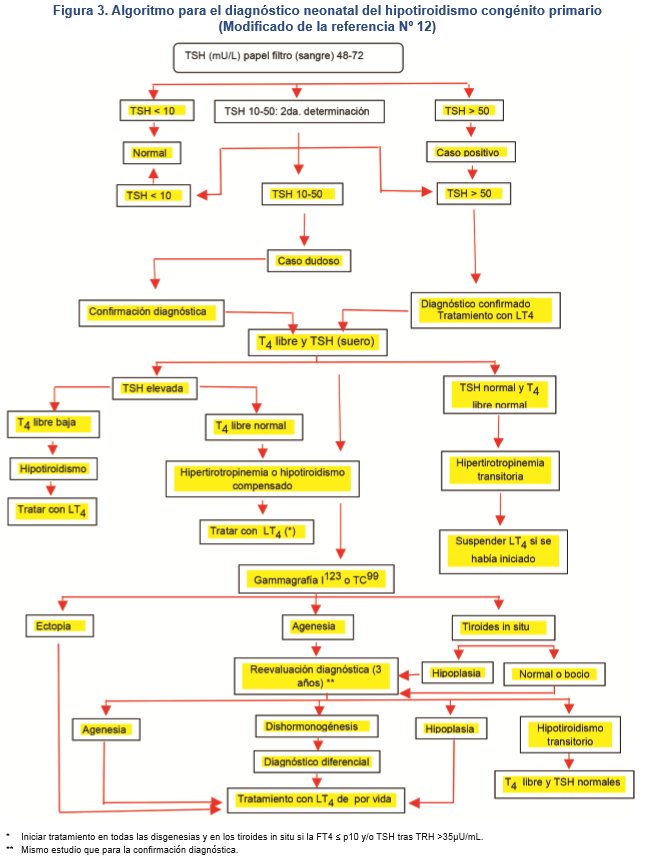
En resumen, se recomienda iniciar el tratamiento de preferencia antes de las dos semanas de vida o inmediatamente después de la prueba sérica confirmatoria: a) a todos los recién nacidos con una concentración capilar en papel filtro de TSH ≥ 40 mU/L, b) si la concentración de T4 libre sérica se halla por debajo del valor normal para la edad, sin considerar la concentración de TSH; y c) si la concentración de TSH venosa es persistentemente > 20 mU/L, aunque la concentración de T4 libre se halle en rango normal.6,10
La concentración de tiroglobulina sérica por debajo del umbral es altamente sugestiva de atiriosis o un defecto completo de la síntesis de tiroglobulina.10
El HC se trata con L-tiroxina (LT4) sódica sintética por vía oral, en dosis única diaria, en ayunas, unos 30 minutos antes de la toma de cualquier alimento para no interferir con su absorción. Las formas de presentación de la LT4 por vía oral son comprimidos que deben ser molidos y diluidos en algunos mililitros de agua o leche materna para ser administrados.10,12 La L-tiroxina puede administrarse en horario matinal o nocturno, pero siempre a la misma hora cada día.10 La absorción suele reducirse con la ingesta de fórmulas infantiles que contienen soya o semilla de algodón, nueces, procesos digestivos en los cuales se reduce la superficie de absorción (intestino corto, cirrosis hepática) y uso de fármacos concomitantes (carbón activado, hidróxido de aluminio, colesteramina, sulfato o gluconato ferroso y propranolol).12
El éxito del tratamiento del hipotiroidismo primario congénito depende -en especial- de su inicio precoz, administración de una dosis inicial y conseguir un correcto equilibrio terapéutico. La dosis inicial adecuada en el RN es aquella que permite normalizar y elevar el nivel de T4 (T4 total > 10 µg/dL; T4 libre > 1.5 ng/dL) lo más rápidamente posible (1 a 2 semanas) y disminuir y normalizar el nivel de TSH a 10 µU/mL o 10mU/L en el primer mes.12
La dosis inicial recomendada por la Academia Americana de Pediatría y la Sociedad Europea de Endocrinología Pediátrica oscila entre 10 a 15 µgl kg/día.9 Así por ejemplo, en un neonato con atiriosis se iniciará el tratamiento a 15 µg/kg/día, a otro con glándula ectópica se iniciará con 12 µg/kg/día y finalmente a un recién nacido con dishormonogénesis se empezará con 10 µg/kg/día.14 Posteriormente, la dosis de mantenimiento de LT4 varía en función de la edad y la gravedad del hipotiroidismo (individualizar a cada paciente).12
- Dosis sugeridas (µg/kg/día), 0-1 mes: 10-15; 1-2 meses: 7-10; 3-5 meses: 4-7; 6-12 meses: 4-6; 1-2 años: 4-6; 3-7 años: 3-4; 7-10 años: 3-4;10-12 años:2-3; >12 años: 2.2,12
- En el control clínico se buscan signos y síntomas sugerentes de infradosificación y supradosificación e incluye la somatometría en cada visita, control de la velocidad de crecimiento, evaluación anual de la edad ósea, densidad mineral ósea y pruebas psicométricas de desarrollo intelectual.12
- El control bioquímico consiste en la monitorización de los niveles de T4 libre y TSH séricos. El primer control se recomienda realizar a las 2 semanas de iniciado el tratamiento, el segundo a las 4 semanas, luego cada 1-2 meses durante el primer semestre de vida,2,12 cada 2-3 meses durante el segundo semestre, cada 3 meses hasta los 3 años, y cada 4 meses con posterioridad.
- Cuando se modifica la dosis en un control, es conveniente realizar una nueva determinación analítica pasadas 4 semanas tras el cambio. Los cambios se suelen hacer aumentando o disminuyendo 12.5 µg de LT4/día.12 Las dosis de LT4 recomendadas son las que permiten mantener el nivel de T4 libre en la mitad superior del rango normal y de TSH entre 0.5 a 2.0 mU/L.2
La rápida normalización de los niveles de hormona tiroidea (dentro de las primeras 2 semanas después de iniciar el tratamiento) y el mantenimiento de concentraciones relativamente altas de T4 libre en el primer año permiten un mejor desarrollo intelectual.15-17 También se requiere la monitorización frecuente de TSH y T4 libre para prevenir la presencia de períodos con niveles suprafisiológicos prolongados de hormona tiroidea.18
En niños con antecedentes de insuficiencia cardiaca debe administrarse el 50% de la dosis correspondiente LT4, incrementando las dosis de acuerdo a los niveles de T4 después de dos semanas.10
Cuando el diagnóstico es tardío o recién se realiza en un lactante con manifestaciones clínicas de hipotiroidismo, el neurodesarrollo es determinado por la severidad de la deficiencia de hormona tiroidea y la duración del hipotiroidismo antes de iniciar el tratamiento. Por ejemplo, un lactante cuyo diagnóstico se realiza a los 3 meses de edad, tendrá un coeficiente intelectual (CI) de 89 (rango 64-107); aquel identificado entre los 3 a 6 meses, su CI futuro será de 71 (rango de 35 a 96); mientras si su diagnóstico fue alrededor de los 6 meses, la media de su CI será de 54 (rango de 25-80).9
En conclusión, podemos señalar que la implementación del programa de detección de HC, introducido hace aproximadamente 40 años ha demostrado ser la mejor estrategia para prevenir el impacto deletéreo de la deficiencia de hormona tiroidea sobre el desarrollo del cerebro en niños afectados y debe ser implementado en todo el mundo para optimizar el cuidado de pacientes con HC tratados en etapas tempranas de la vida y efectuar un regular seguimiento médico y control apropiado de su tratamiento.
Referencias
1. Rastogi MV, LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis 2010;17:1-22. [ Links ]
2. American Academy of Pediatrics. Update of new-born screening and therapy for congenital hypothyroidism. Pediatrics 2006;117:2290-2303. [ Links ]
3. Léger J. Congenital hypothyroidism: a clinical update of long term outcome in young adults. Eur Soc Endocrinol 2015;172:67-77. [ Links ]
4. Almandoz JP, Gharib H. Htpothyroidism; etiology, dianosis, and management. Med Clin North Am 2012;96:203-221. [ Links ]
5. Castanet M, Lyonnet S, Bonaiti-Pellie C, Polak M, Czernichow P and Leger J. Familial forms of thyroid dysgenesis among infants with congenital hypothyroidism. N Engl J Med 2000;343:441-2.
6. Counts D, Varma SD. Hypothyroidism in children. PediatrRev 2009;7:251-8. [ Links ]
7. Salim FA, Surendra KV. Congenital hypothyroidism and the importance of universal newborn screening. Indian J Pediatr 2014;81:53-7. [ Links ]
8. Harris KB, Pass KA: Increase in congenital hypothyroidism in New York State and in the United States. Mol GenetMetab 2007; 91(3):268-277. [ Links ]
9. Ford G, LaFranchi SH. Screening for congenital hypothyroidism: a worldwide view of strategies. Best Pract Res Clin Endocrinol Metab 2014;28:175-87. [ Links ]
10. Léger J, Olivieri A, Donaldson M. Torresani T, Krude H, Vliet GV, et al. European society for paediatric endocrinology consensus guidelines on screening, diagnosis and management of congenital hypothyroidism. J Clin Endocrinol Metab 2014;99: 1-22. [ Links ]
11. Bursell JD. Interpretation of thyroid function in children. Pediatr Child Health 2007:17:361-6. [ Links ]
12. Argente J, Soriano L. Patología de la glándula tiroides: hipotiroidismo, hipertiroidismo, nódulo de tiroides y cáncer de tiroides. En: Argente J, Soriano L, ed. Manual de endocrinología pediátrica. 1ra. ed. Madrid: Ergon.,2010:109-148.
13. Grüters A, Krude H. Detection and treatment of congenital hypothyroidism. Nat Rev Endocrinol. 2012;8:104-113. [ Links ]
14. Mathai S, Cutfield WS, Gunn AJ. A novel therapeutic paradigm to treat congenital hypothyroidism. Clin Endocrinol(Oxf)2008;69: 142-47. [ Links ]
15. Selva KA, Harper A, Downs A, Blasco PA,Lafranchi SH: Neurodevelopmental outcomes in congenital hypothyroidism: comparison of initial T4 dose and time to reach target T4 and TSH. J Pediatr 2005; 147: 775-80. [ Links ]
16. Selva KA, Mandel SH, Rien L, et al: Initial treatment dose of L-thyroxine in congenital hypothyroidism. J Pediatr 2002; 141:786-92. [ Links ]
17. Heyerdahl S, Oerbeck B: Congenital hypothyroidism: developmental outcome in relation to levothyroxine treatment variables. Thyroid 2003; 13: 1029-1038. [ Links ]
18. Balhara B, Misra M, Levitsky LL: Clinical monitoring guidelines for congenital hypothyroidism: laboratory outcome data in the first year of life. J Pediatr 2011; 158: 532-37. [ Links ]

