










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO Compartir

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
versión On-line ISSN 1024-0675
Rev. bol. ped. vol.54 no.2 La Paz 2015
CASO CLÍNICO
Deficiencia congénita de Proteína C asociada a polimorfismo C677T del gen de la 5,10-metiltetrahidrofolato reductasa, a propósito de un caso
Congenital deficiency of protein C associated with C677T polymorphism of the gene from the 5,10-methyltetrahydrofolate reductase. Case report
Drs.: Carla Varela Morales*, Julio Álvarez Endara**, Manuel Pantoja Ludueña***, Eduardo Mazzi Gonzales de Prada****
* Médico Residente 3o Año. Hospital del Niño Dr. Ovidio Aliaga Uría. La Paz, Bolivia.
** Hematólogo - Hemoterapeúta. Hospital del Niño Dr. Ovidio Aliaga Uría. La Paz, Bolivia.
*** Jefe de Servicio de Neonatología. Hospital del Niño Dr. Ovidio Aliaga Uría. La Paz, Bolivia.
**** Perinatólogo. Hospital del Niño Dr. Ovidio Aliaga Uría. La Paz, Bolivia.
Correspondencia: Dr. Manuel Pantoja. Correo electrónico: mpantoja@acelerate.com
Conflicto de intereses: el presente trabajo no tiene conflicto de intereses.
Artículo aceptado para su publicación el 1/06/15.
Resumen
Se presenta el caso clínico de un recién nacido con trombofilia hereditaria por deficiencia congénita de proteína C asociada a polimorfismo C677T del gen de la 5,10-metiltetrahidrofolato reductasa.
Palabras clave:
Rev Soc Bol Ped 2015; 54 (2): 72-6: trombofilia hereditaria, deficiencia congénita de proteína C.
Abstract
We present a clinical case of a newborn with inherited thrombophilia by congenital deficiency of protein C associated with the gene from the 5, 10-methyltetrahydrofolate reductase C677T polymorphism.
Key words:
Rev Soc Bol Ped 2015; 54 (2): 72-6: inherited thrombophilia, congenital deficiency of protein C.
Introducción
La hemostasia es el proceso activo que coagula la sangre en las zonas donde se haya producido una lesión de los vasos sanguíneos; a la vez que limita el tamaño del coagulo únicamente al área lesionada. Si la coagulación es defectuosa se producirá una hemorragia. Si por el contrario es excesiva, se producirá complicaciones trombóticas. Los componentes principales del proceso hemostático son las paredes vasculares, las proteínas de coagulación, las proteínas anticoagulantes y el sistema fibrinolítico1.
El endotelio vascular intacto es la primera barrera frente a la hemorragia. Las células endoteliales que tapizan la pared vascular normalmente inhiben la coagulación; cuando se produce una lesión vascular, se desencadena una vasoconstricción y el flujo de sangre contacta con la matriz subendotelial. El factor de von Willebrand (FvW) entra en contacto con las proteínas de la matriz subendotelial, cambiando su configuración y proporcionando el pegamento al que se unen los receptores de FvW de las plaquetas. Una vez adheridas las plaquetas se activan y liberan los gránulos que contienen difosfato de adenosina (ADP), tromboxano A2 y otras proteínas almacenadas. Este proceso desencadena la agregación y la atracción de otras plaquetas para formar el coagulo plaquetario. La agregación implica una interacción de receptores específicos de la superficie de las plaquetas con las proteínas hemostáticas del plasma, en especial el fibrinógeno1.
Una de las proteínas de la matriz subendotelial que queda expuesta tras la lesión vascular es el factor tisular. Inmediatamente tras la unión entre las proteínas de la matriz subendotelial expuestas y el FvW, también se libera el factor tisular, que se une al factor VII y activa la cascada de la coagulación como se expone en la figura # 1. En la cascada, los factores de la coagulación inactivos, designados con números, se activan. El factor de coagulación activado inicia la activación del siguiente factor de la coagulación de una manera sucesiva y sistemática1.
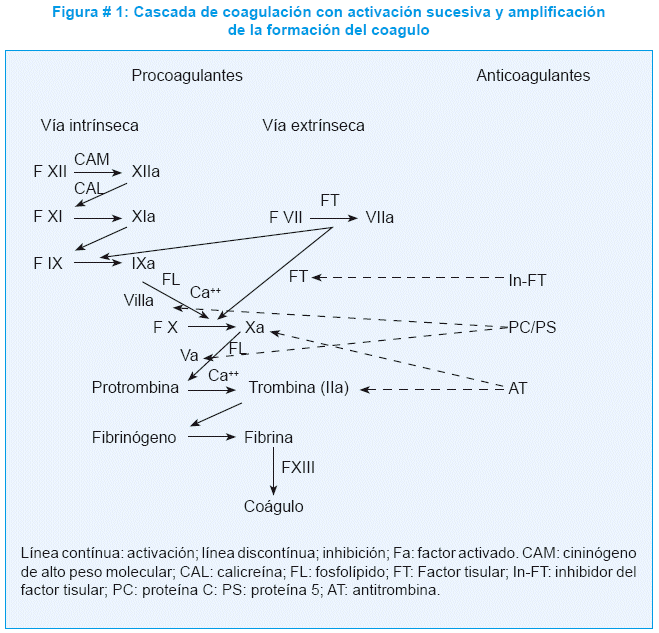
La proteína C es una proteína inhibidora plasmática que, una vez activada, inhibe la formación del coágulo y favorece la fibrinólisis. Es un factor clave en la regulación de la coagulación, se sintetiza en el hígado y es dependiente de la vitamina K. Se convierte en proteína C activa por la acción del complejo trombina-trombomodulina sobre la superficie celular endotelial; activa a un activador del plasminógeno, lo que determina aumento de la fibrinólisis y con la proteína S como cofactor, produce la proteólisis limitada de los factores Va y VIIIa1,2. La deficiencia de proteína C es una alteración autosómica dominante con penetrancia variable, que se localiza en el cromosoma 2, su expresión genética puede ser homocigota o heterocigota y su prevalencia es muy variable, tanto en controles sanos como en pacientes con trombosis3,4.
La enfermedad tromboembólica es considerada como una enfermedad multifactorial. Es necesaria la presencia de factores genéticos, que predisponen al individuo a la trombosis, y factores ambientales, que desencadenan la trombosis (interacción gen-ambiente)3.
El hecho de que la trombosis pudiera ser un rasgo hereditario no se reconoció hasta 1965, año en el que se describe el déficit de antitrombina. Posteriormente, se describieron el déficit de proteína C en 1981, el déficit de proteína S en 1984, la mutación factor V Leiden en 1994 y la mutación G20210A del gen de la protrombina en 1996. En 1995 Frost et al describieron un nuevo gen candidato para el riesgo trombótico, la mutación C677T del gen de la 5,10-metilentetrahidrofolato reductasa (C677T MTHFR)3.
La mutación C677T MTHFR es un hallazgo frecuente en la población caucasiana, encontrándose hasta en el 38% de los sujetos no seleccionados, pero es poco frecuente en raza negra, nativos de América del Sur y aborígenes de Oceanía3,4.
Caso clínico
Paciente de 12 días de vida de sexo femenino procedente y residente de la ciudad de Potosí, que ingresa por un cuadro clínico de 7 días de evolución caracterizado por hipoactividad, rechazo del seno materno, llanto débil, tres días después la madre nota coloración violácea de región glútea izquierda, además de la presencia de lesiones en región de muslo con mayor compromiso del pie ipsilateral, por lo que es internada en el Hospital Bracamonte de Potosí, donde se estabiliza con aporte de soluciones parenterales y oxígeno permaneciendo internada por lapso de 24 horas, ante la falta de mejoría del estado general y de la perfusión distal del pie izquierdo, con hipotermia local y coloración negruzca, se decide su transferencia al Hospital Santa Barbara de Sucre, donde se realiza la valoración con Cirugía Cardiovascular, completando una ecografía doppler que evidencia disminución del flujo sanguíneo pedio izquierdo iniciando la administración de enoxaparina y finalmente es transferida a nuestro institución Hospital del Niño Dr. Ovidio Aliaga.
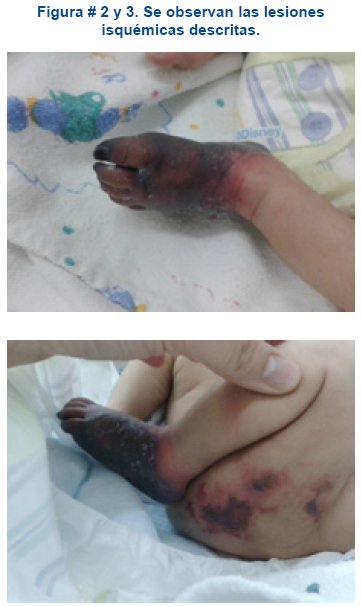
Al examen físico paciente estable hemodinamicamente, sin sintomatología respiratoria ni neurológica; llamaba la atención a nivel de pie izquierdo coloración negra de la piel, dura de aspecto momificado, bien delimitado por debajo de la articulación bimaleolar, frio al tacto y ausencia de pulso pedio, así mismo se aprecia a nivel de muslo lesiones violáceas, con presencia de flictenas, que se extienden desde glúteo hasta tercio inferior del muslo, acompañado de aumento de volumen hasta tercio medio de pierna, ver figuras # 2 y 3.
Valorado por cirugía cardiovascular, que realiza una nueva ecografía doppler que indica disminución del flujo sanguíneo a nivel fémoro-poplíteo y ausente en pedio izquierdo. Se mantiene la enoxaparina, y antibióticos de amplio espectro. Se inicia protocolo de estudio para trombofilia, ya contando con los resultados de laboratorios reportan: Hto: 40%, Hb: 13.2 g/dL, TP: 76%, GB: 11.200/mm3, PCR: 12.5 mg/dL, Proteína C: 12% Proteína S: 56%, Antitrombina III 46mg%, perfil inmunológico contando con ANA, anti DNA, FR, y anticuerpos antifosfolípidos dentro de parámetros normales; así mismo se completa la toma de muestra para biopsia de lesiones glúteas que reportan fascitis necrosante.
Se realiza dosificación de Proteína C a la familia de la paciente, reportando en el padre cifra de 62%, en la madre 58% y en el hermano mayor 72%; considerando que los valores normales se encuentra en el rango de 60% a 100%, por estos datos se completa estudio genético en búsqueda de la mutación del gen 5,10-metiltetrahidrofolato reductasa tanto a la paciente como a la familia, encontrándose en el padre polimorfismo heterocigoto del gen C677T (portador sano), en la madre polimorfismo homocigoto del gen C677T (portadora sana), hermano menor portador del polimorfismo heterocigoto del gen C677T (portador sano) y en la paciente polimorfismo homocigoto del gen C677T.
Se realiza una junta médica para realizar la ablación del píe izquierdo, se mantiene la administración de enoxaparina hasta nuevo control de dosificación de proteína C.
Discusión
La frecuencia de trombofilia hereditaria en la población mundial es de1:25000 a 1:50000 debido las propiedades especiales del sistema hemostático en la niñez y la infancia, la trombofilia hereditaria solo se presenta sintomáticamente en el 5%5.
Se han descrito al menos 161 diferentes mutaciones responsables de este déficit, el cual es de herencia autosómica dominante. Es importante destacar que la presentación homocigota es muy rara y esta situación se asocia a graves eventos trombóticos de aparición neonatal.
Este desorden inicialmente descrito por Dahlback en 1993, actualmente es considerado como la causa más importante de tromboembolismo en adultos y niños5,6.
El paciente tuvo manifestaciones clínicas dentro de los primeros 12 días, con cambios en la coloración del miembro inferior izquierdo, se realizo el protocolo para trombofilia y se determino que el paciente tenía una deficiencia de proteína C3,5,6.
Se realizaron exámenes en los padres y se determino que la madre presentaba también una deficiencia de proteína C, a diferencia de los pacientes homo-cigotos, los heterocigotos presentan una clínica diferente a una purpura fulminante descrita en la literatura como, trombosis, manifestaciones cutáneas y renales, y trastornos retinianos. Se describió un caso similar de un niño con trombosis en miembro inferior derecho en la Gaceta de México con un déficit de proteína C7,8,9.
A pesar que se realizo la amputación en nuestro caso y en el descrito en México por, el diagnostico temprano y el tratamiento multidisciplinario realizado en el Hospital del Niño Ovidio Aliaga por las especialidades de Neonatología, Pediatría, Traumatología, Hematología y seguir un protocolo adecuado para estudio de coagulopatías el pronóstico es bueno, se lo gro salvar la vida del paciente y actualmente se halla en controles por consultorio externo donde se tratara de mejorar la calidad de vida.
Referencias
1. Lemus M, Arríaga J, Salinas M. Deficiencia congénita de proteína C. Informe de un caso. Gac Méd Méx 2005;141:229-33. [ Links ]
2. González JR, Pérez E, Alberca I, Lozano FS. Influencia de la mutación C677T del gen de la metilentetra hidrofolato reductasa en la enfermedad tromboembólica venosa. Angiología 2010;62:225-31 [ Links ]
3. Zamora L, Agramonte O, Rodríguez L. Deficiencia de proteínas C y S: marcadores de riesgo trombótico. Rev Hematol 2013;26:1-7. [ Links ]
4. Aversa L. Trombosis en Pediatría: su relación con el síndrome antifosfolipídico. Arch Argent Pediatr 2001; 99:293-5. [ Links ]
5. Tripodi A, Mannucci P. Laboratory investigation of thrombophilia. Clin Chem 2001;47:1597-606. [ Links ]
6. Muwakkit SA, Majdalani M, Hourani R, Mahfouz RA, Otrock ZK, Bilalian C, et al. Inherited thrombophilia in childhood arterial stroke: data from Lebanon. Pediatr Neurol 2011;45:155-8. [ Links ]
7. Uri Seligssohn, Aarón Lubetsky: Genetic susceptibility to venous trombosis. N Engl J Med 2001;344:1222-31. [ Links ]
8. Dahlback B, Carisson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA 1993;90:1004-8. [ Links ]

