










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
On-line version ISSN 1024-0675
Rev. bol. ped. vol.53 no.1 La Paz 2014
CASO CLÍNICO
Diagnóstico clínico y molecular de síndrome de Rett. A propósito de un caso
Clinical and molecular diagnosis of Rett's syndrome. A case report
Drs.: Beatriz Luna Barrón*, José Liders Burgos Zuleta**, Gonzalo Taboada López***, Ana Rada Tarifa****, Erika Lafuente Álvarez****
* Médico Genetista. Docente Investigador. Instituto de Genética. Universidad Mayor de San Andrés.
** Médico Imagenólogo. Caja de Salud de la Banca Privada. Centro de Imagen Médica Avanzada
*** Médico Genetista. Director Instituto de Genética. Universidad Mayor de San Andrés
****Bioquímica Genetista. Asistente Investigación I. Instituto de Genética. Universidad Mayor San Andrés.
Correspondencia: Dra. Beatriz Luna Barrón. Correo electrónico: blunab3@gmail.com
Conflicto de intereses: los autores expresan que el presente trabajo no tiene conflicto de intereses.
Artículo aceptado para su publicación el 04/03/14.
Resumen
Describimos el caso de una niña de 2 años de edad, referida al servicio de genética con retraso del desarrollo psicomotor, regresión neurológica importante, microcefalia y conducta estereotipada. El estudio molecular realizado reveló el estado heterocigoto de una mutación de sentido erróneo en el exón 4 del gen responsable de este síndrome (análisis mediante secuenciación). Las características clínicas de esta enfermedad sugieren un patrón de desarrollo anormal de la corteza cerebral en la infancia tardía relacionada con la disfunción de la proteína MECP2. Es una importante causa genética de autismo y debe ser considerada en casos de pérdida progresiva de habilidades adquiridas.
Palabras clave:
Rev Soc Bol Ped 2014; 53 (1): 8-11: Síndrome de Rett, mutación, autismo.
Abstract
We describe a case of two year old child referred to the service of Genetics with psychomotor retardation, severe neurological regression, microcephaly and stereotyped behavior. The molecular study revealed the heterozygous state of a missense mutation in exon 4 of the gene responsible for this syndrome (sequencing analysis). The clinical features of the disease suggest a pattern of abnormal development of the cerebral cortex in late childhood related to the dysfunction of the MECP2 protein. It's a major genetic cause of autism and should be considered in cases of progressive loss of acquired skills.
Key words:
Rev Soc Bol Ped 2014; 53 (1): 8-11: Rett's Syndrome, mutation, autism.
Introducción
El síndrome de Rett (SR) es una enfermedad del neurodesarrollo, con regresión de habilidades de las áreas del lenguaje y motoras. Se presenta en 1 de cada 8300 a 15000 mujeres1. Es una de las causas genéticas del Trastorno del Espectro Autista. El gen implicado es MECP2 (80% de los casos de la forma clásica) localizado en el cromosoma X región q28, este gen codifica para la proteína methil-CpG-binding protein 21,2. El 99% de los casos son mutaciones de "novo" (casos únicos en la familia). Afecta casi exclusivamente al sexo femenino, las mujeres heterocigotas para la mutación padecen el síndrome demostrando el carácter dominante de la enfermedad (ligada al X dominante)3. Sin embargo, también se ha descrito en el sexo masculino cuando se asocia a mosaicismo o alteraciones gonosómicas (tipo síndrome de Klinefelter)1,3,4.
Caso clínico
Se trata de una paciente de sexo femenino, producto de segunda gestación, padres no consanguíneos. Caso único en la familia. Sin datos de exposición a teratógenos durante la gestación, ni factores adversos en período prenatal, perinatal ni postnatal. Nació a las 40 semanas de gestación, parto eutócico, sin complicaciones, peso al nacimiento: 3500 g (p50), talla 51 cm (p50), perímetro cefálico 34 cm (p50). Apgar 9/10. Desarrollo psicomotor normal acorde a la edad hasta los 8 meses (pronunciaba monosílabos, habilidades motoras con las manos como agarrar la cuchara, señalar objetos, etc,).
Sin embargo, a partir de los 9 a 12 meses los padres observaron irritabilidad continua, retraso del desarrollo psicomotor (no gateaba, no caminaba, pronunciaba pocas palabras), pérdida de habilidades con las manos (ya no tomaba objetos con las manos), comenzó a presentar movimientos estereotipados con las manos y bruxismo continuo. El motivo de referencia a la especialidad de Genética fue trastorno del espectro autista. A los 2 años la paciente continúo con la pérdida de habilidades adquiridas, retraso psicomotor marcado, bruxismo, llamaban la atención los movimientos continuos con las manos (frote de manos), episodios de llanto inconsolable. A la exploración física perímetro cefálico 38 cm (< p5) microcefalia, resto sin dismorfias faciales ni corporales. Exámenes de laboratorio de rutina (hemograma, bioquímica sanguínea, examen general de orina) sin alteraciones significativas, panel metabólico sin alteraciones. Cariotipo en sangre periférica con bandeo G: 46,XX en 20 metafases. Tomografía de cerebro: Sin alteraciones evidentes por el método de estudio.
Por los antecedentes clínicos (sexo femenino con periodo perinatal normal, retraso de desarrollo psicomotor, pérdida paulatina de habilidades adquiridas, microcefalia postnatal, bruxismo permanente, irritabilidad, etc.) se sospechó de SR, motivo por el cual previa autorización de los padres se solicitó estudio molecular del gen MECP2 en busca de mutaciones patológicas.
El estudio se realizó con una muestra de sangre periférica de la paciente de la cual se extrajo ADN mediante procedimiento de precipitación salina y cuantificación del mismo, posteriormente se procedió a la amplificación por PCR (reacción en cadena de la polimerasa) y secuenciación bidireccional del total de la región codificante del gen MECP2 (Methyl CpG binding protein 2) y regiones intrónicas adyacentes. El resultado mostró la presencia de heterocigosis de la mutación c.473C>T p. Thr 158 Met en el exón 4 del gen estudiado responsable del SR, confirmando dicho diagnóstico. Ver figura # 1.
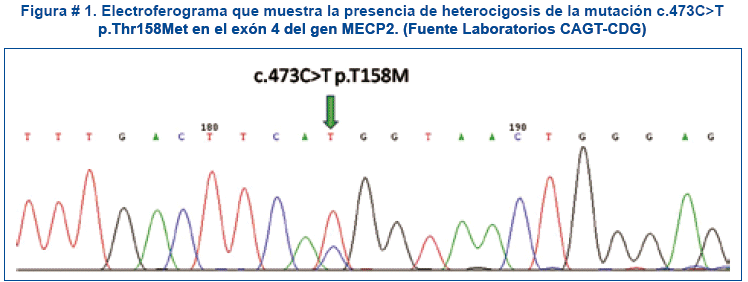
Se procedió a dar asesoramiento genético a ambos padres, explicando la causa genética, el modo de herencia ligado al X dominante y la posibilidad de evitar complicaciones secundarias al diagnóstico (tratamiento sintomático adecuado, así como una terapia educacional adaptada), con anticonvulsivantes, manejo nutricional y terapia de rehabilitación.
Discusión
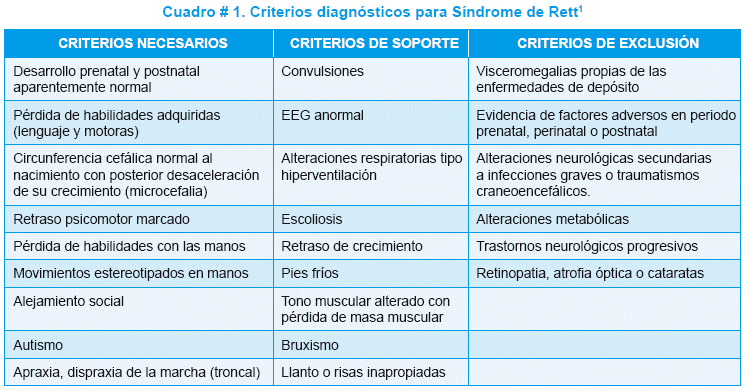
Esta enfermedad, presenta un amplio espectro de manifestaciones clínicas (Cuadro #1), tales como alteración del desarrollo con retraso del mismo y pérdida progresiva de habilidades adquiridas, movimientos estereotipados (sobre todo en las manos), microcefalia, crisis convulsivas y retraso mental5-6. Además se han descrito 4 estadíos clínicos (Cuadro # 2).

Además del síndrome de Rett clásico, la literatura reporta otras variantes: como la variante con crisis de presentación temprana, la variante Zapella, la variante congénita y la de regresión tardía2. En estos casos los genes implicados son CDKL5 y FOXG3,4.
El SR se describe como la segunda causa de retraso mental en el sexo femenino, precedido en frecuencia por el síndrome de Down y en una causa importante de TAE1 de tipo complejo, por lo que éste diagnóstico debe ser considerado en casos de sexo femenino con retraso psicomotor, regresión neurológica y autismo.
Su heterogeneidad fenotípica se relaciona con la inactivación al azar del cromosoma X2-11.
Las características clínicas de esta enfermedad sugieren un patrón de desarrollo anormal de la corteza cerebral en la infancia tardía4,5 relacionadas con la disfunción de la proteína MECP2, Esta proteína es una molécula de unión al RNA (ácido ribonucléico) relacionada con el splicing alternativo (corte y empalme de RNA mensajero) en el cerebro1,4. Su expresión muestra niveles elevados durante la maduración neuronal, se requiere su presencia para la modulación de la sinapsis dendrítica1,3 y es importante para la función de otros genes.
La patología molecular explica porque un cambio genético determinado da por resultado un fenotipo clínico en particular, como en el caso de la paciente. En el SR existe heterogeneidad alélica (capacidad de un gen de causar el mismo fenotipo por distintas mutaciones)5, es decir, en el gen MECP2 se han descrito aproximadamente 20 mutaciones diferentes en pacientes con SR. Así mismo, tiene heterogeneidad de locus al existir variantes en otros genes involucrados.
La mayoría de las mutaciones encontradas en el gen MECP2 corresponden a alteraciones en la secuencia (80%)1,4, el 8% corresponde a una deleción (pérdida de material) o duplicación (ganancia de material) de un segmento del gen5. La mutación encontrada en la paciente fue una alteración en la secuencia: c.473C>T p. Thr 158 Met correspondiente a una "mutación de sentido erróneo"7, que consisten en una sustitución en la cadena de DNA de la paciente en el nucleótido 473 de citocina (normal) por Timina (anormal) lo que corresponde en la proteína final (MECP2) a un cambio del amninoácido treonina por metionina, modificando la función. Esta mutación esta descrita entre las más frecuentes en la literatura10.
Si bien el SR no tiene un tratamiento curativo, investigadores han demostrado la utilidad de la mecasermina8 (Factor recombinante humano IGF-1) como fármaco neuromodulador que permite una mejoría importante de la esfera neuroconductual, con buena tolerancia en las pacientes.
Referencias
1. Del Castillo V. Síndrome de Rett. En: Del Castillo V, Uranga D, Zafra G. Eds. Genética Clínica.1era ed. Mexico D.F; Manual Moderno.2012.p.203-7. [ Links ]
2. Firth Helen V, Hurst Jane A, Hall Judith G. Eds. Oxford Desk Reference Clinical Genetics. 3era ed. U.K.; Oxford University Press. 2006.p.408-9.
3. Christodoulou J, Ho G. MECP2-Related Disorders. Gene Reviews/publicación periódica en línea/2014/ fecha de consulta 2014 enero 30. Localizable en http://www.ncbi.nlm.nih.gov/books/NBK1497/
4. Lapunzina P. Sindrome de Rett. En: Delgado A, Galán E, Guillén-Navarro E, Penchaszadeh V, Romeo C, et al. Eds. Asesoramiento Genético en la Práctica Médica. 1era Ed. Madrid, España; Médica Panamericana. 2012.p.211-37. [ Links ]
5. Nussbaum R, Mclnnes R, Williard H, Eds. Genética en Medicina. 5a ed. Barcelona. España; Masson. 2004.p. 143-6.
6. Jones KL. Ed. Smith's Recognizable Patterns of Human Malformation. 6ta ed. Seatle USA. Elsevier. 2006.p.345
7. Strachan J. Ed. Genética Humana. 3era ed. EEUU. Mc Graw Hill. 2006.p.706
8. Khwaja O, Ho E, Barnes K, O'Leary H. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci 2014;111:4596-601. [ Links ]
9. Chen H.Ed. Atlas of Genetic Diagnostic and Counseling. 1era Ed. USA; Humana Press Inc. 2006.p.839-3.
10. Smeets E, Chenault M, Curfs L, Schrander-Stumpel C, Frijns JP. Rett syndrome and long-term disorder profile. Am J Med Genet Part A 2009; 149:199-205. [ Links ]
11. Renieri A, Meloni I, Longo I, Ariani F, Mari F, Pescucci C, Cambi F. Rett syndrome: the complex nature of a monogenic disease. J Mol Med (Berl) 2003 ;81:346-54. [ Links ]

