










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
versión On-line ISSN 1024-0675
Rev. bol. ped. v.46 n.1 La Paz 2007
CASO CLINICO
22q11 deletion syndrome, case presentation
Drs.: Pablo Mattos Navarro*, Igor Salvatierra Frontalilla**, Andrés Bartos Miklos***
* Médico Pediatra, Unidad de Terapia Intensiva pediátrica, Hospital Materno Infantil (HMI)
** Médico Genetista
*** Médico Pediatra Neonatólogo, Profesor de Pediatría, UMSA, Director medico, Hospital Materno Infantil
Resumen
Los síndromes de DiGeorge y Velocardiofacial son entidades clínicas incluidas en el síndrome de deleción 22q11, la alteración genómica más frecuente encontrada en humanos, 1 por cada 4000 nacimientos. Por una parte incluye fenotipos extremos que no presentan dificultad en su identificación como también formas intermedias que complican su diagnóstico clínico. Presentamos el caso de un lactante menor de tales características, describimos el proceso mediante el cual llegamos a un diagnóstico presuntivo orientados por las manifestaciones multisistémicas, su manejo, pronóstico y una breve revisión de la literatura.
Palabras claves:
Rev. Soc. Bol. Ped. 2007; 46 (1): 24-8: malformaciones congenitas, Síndrome de DiGeorge, Síndrome de velo cardiofacial, síndrome de deleción 22q11
Abstract
DiGeorge syndrome and Velo-cardio-facial syndrome are clinical manifestations of 22q11 deletion syndrome, the most common genomic alteration found in humans, 1 for every 4000 births. On one side it includes extreme phenotypes that are easily identifiable, but some intermediate forms are difficult to recognize. We present the case of an infant that presented with features compatible with these syndromes, we describe the process that lead us to the diagnosis, the prognosis, and we present a brief revision of the literature.
Rev. Soc. Bol. Ped. 2007; 46 (1): 24-8: congenital malformations, DiGeorge syndrome, velo-cardio-facial syndrome, 22q11 deletion syndrome
El síndrome de deleción 22q11 es un cuadro de anomalía del desarrollo caracterizado por una haploinsuficiencia de una región genómica por deleción de tres megabases en el cromosoma 22q11 y se asocia a una variedad de fenotipos clínicos que incluyen el Síndrome de DiGeorge, el Síndrome velocardiofacial, la anomalía facial conotruncal y defectos cardíacos congénitos esporádicos o familiares2,3.
En el humano la deleción cromosómica 22q11 es la deleción intersticial más frecuentemente encontrada , de las cuales entre 5-10% son hereditarias. La deleción ha sido asociada a malformaciones congénitas variables que comprometen diferentes órganos, con severidad y expresión variables5.
La incidencia estimada es mayor de 1 en 4.000 nacimientos4 y se ha descrito la microdeleción intersticial 22q11 en un 5% de los recién nacidos con defectos cardíacos. Las cardiopatías congénitas que se observan son defectos del desarrollo del cono-truncal, siendo las formas más frecuentes la tetralogía de Fallot, el arco aórtico interrumpido y el tronco arterioso, así como la comunicación interventricular con y sin estenosis de la válvula pulmonar6. El cuadro es más amplio, observándose también anomalías otorrinolaringológicas, genitourinarias, de la función paratiroidea, inmunológicas y neurológicas.
La región 22q11 contiene genes que juegan un papel importante, durante la embriogénesis, en el desarrollo normal de los órganos derivados de la tercera y cuarta bolsa faríngea, el timo, las glándulas paratiroideas y la estructura conotruncal del corazón4. Entre estos genes se incluyen algunos factores de trascripción involucrados en el desarrollo cardíaco7,8 El fenotipo más grave lo constituye el síndrome de DiGeorge, descrito en 1965, que incluye la implantación baja de orejas, filtrum corto, telecantus con fisuras palpebrales cortas, hipocalcemia por hipoplasia paratiroidea, defectos en el tracto de salida del corazón (ej., tetralogía de Fallot) y déficit inmunológico, principalmente de células T por hipoplasia del timo8. El Síndrome Velocardiofacial, conocido también como Síndrome de Shprintzen, combina anomalías cardíacas con una apariencia facial inusual, paladar hendido -a menudo submucoso-, baja talla y dedos largos, delgados e hiperextensibles. La facies se caracteriza por una nariz prominente, bulosa y alas nasales hipoplásicas, micrognatia, microcefalia y ocasionalmente anomalías oculares. El retardo mental, definido como un coeficiente intelectual menor a 70, está presente en un 50% de los casos y es usualmente leve9. Los rasgos clínicos pueden ser muy variables10. Existe un considerable sobrelapamiento con el Síndrome de DiGeorge, la mayoría de los pacientes tienen la deleción 22q11 a nivel molecular11,12. La importancia de una correcta fenotipificación de los afectados con la deleción 22q11 radica en el pronóstico resultante, dado que en el caso más severo, el Síndrome de DiGeorge, la sobrevida puede estar seriamente comprometida por la hipocalcemia de difícil corrección y un compromiso inmunológico importante que se complica con infecciones severas; mientras que en el Síndrome Velocardiofacial el cuadro clínico puede ser menos severo. La dificultad en la fenotipificación se hace evidente cuando se presentan las formas sobrelapadas y no se puede establecer con facilidad el síndrome correspondiente, tal como presentamos en el presente caso.
Recién nacido de sexo masculino, procedente de Caranavi, producto de séptimo embarazo, parto por cesárea iterativa, con llanto inmediato e historia negativa para hipoxia, 2900 g de peso al nacimiento y alta hospitalaria al tercer día de vida. Presentó en su domicilio cianosis, vómitos, alzas térmicas, hiporexia e hipoactividad. Fue hospitalizado en Caranavi y se inició antibioticoterapia (ampicilinagentamicina), por considerarse un cuadro de sepsis y una conjuntivitis bacteriana aguda, sin respuesta favorable al tratamiento, por lo que fue remitido a nuestro hospital.
Ingresó en mal estado general, al cuadro anterior se agregó estridor laríngeo, crisis de apneas y requirió ventilación mecánica. Las apneas remitieron; sin embargo presentó abundantes secreciones y posteriormente se agregaron crisis convulsivas tónicoclónicas generalizadas, con un EEG patológico que reveló ondas lentas focalizadas, adicionándose a su tratamiento anticonvuslivantes (fenobarbital, ácido valproico y diazepam).
A los dos meses de edad presentó el siguiente fenotipo: baja talla proporcional (-2DS), perímetro cefálico adecuado para la talla, plagiocefalia sin cráneosinostosis, fisuras palpebrales oblicuas hacia abajo, baja implantación anterior del cabello, narinas hipoplásicas, orejas displásicas, rotadas hacia atrás, paladar ojival íntegro, micrognatia relativa, voz nasal, hernia umbilical, pulgar aducto bilateral, aracnodactilia en manos y pies, rigidez muscular con relativa hiperreflexia (Figura # 1).
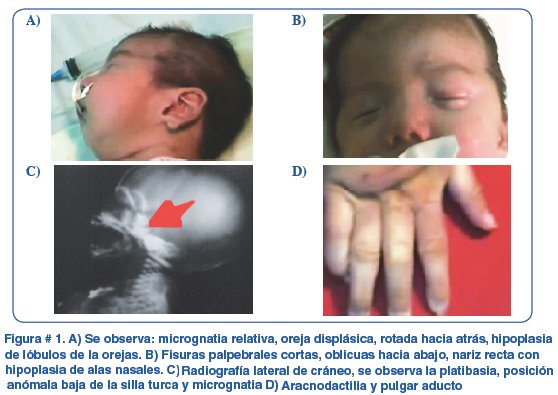
Se encontró lo siguiente con los exámenes de gabinete: ecocardiografia: persistencia del conducto arterioso, TAC de cerebro con atrofia cortical, TAC de tórax con adenomegalias en mediastino, ecografía renal y de vías urinarias dentro de parámetros normales, hormonas tiroideas dentro de limites para la edad, hipoparatiroidismo, inmunoglobulinas G,A y M dentro de limites para la edad, hipocalcemia e hipercalciuria.
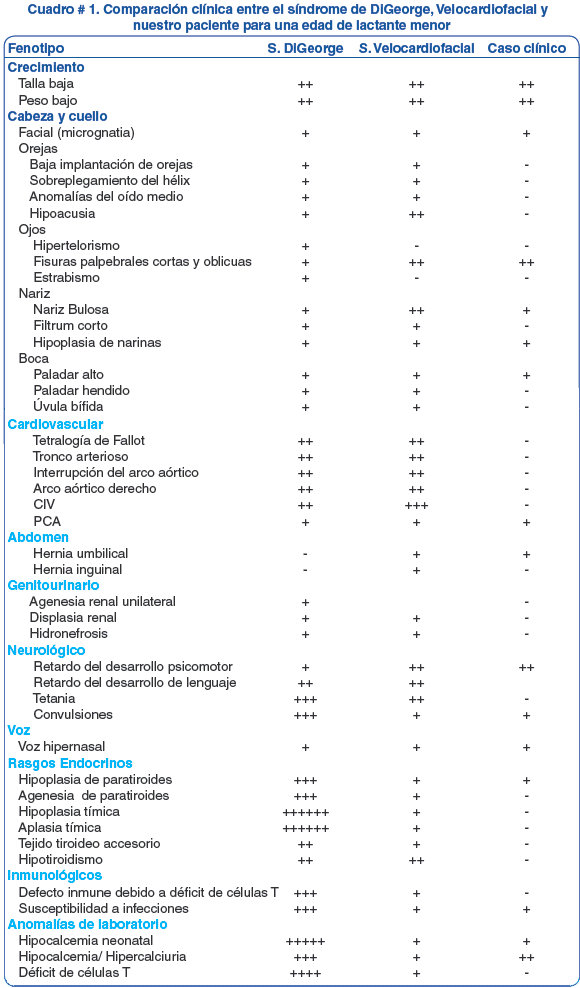
El fenotipo particular del paciente más los hallazgos de gabinete sugirió un posible síndrome genético (cuadro # 1), hecho que requirió el concurso multidisciplinario de pediatras, endocrinólogos, cirujanos maxilofaciales y genetista, hasta determinar el diagnóstico de síndrome de deleción 22q11 con un fenotipo sobrelapado entre el Síndrome de DiGeorge y el Síndrome de Shprintzen.
Discusión
El síndrome de deleción 22q11 es una anomalía descrita en todas las poblaciones del mundo, por ello debemos considerar posible su presencia entre nuestros neonatos, infantes, pre-escolares, púberes e incluso adultos, dado que el espectro de manifestaciones es extremadamente variable. La identificación temprana es la base fundamental para lograr un abordaje oportuno y para ello la hipocalcemia, las cardiopatías complejas o las facies anormales asociadas con múltiples malformaciones deberán ser los signos de alerta para la sospecha temprana del síndrome de deleción 22q11. Una correcta semiología para la rápida identificación de malformaciones mayores y menores deberá ser sustentada por unadecuado entrenamiento de un equipo multidisciplinario. Si consideramos la frecuencia de la deleción 22q11 deberíamos esperar en La Paz alrededor de 12 casos por año y a nivel nacional alrededor de 18 casos anuales, lo cual significa que es tiempo de instaurar protocolos de diagnóstico por citogenética molecular (FISH) o bien alternativas por marcadores STRs que son las únicas pruebas que confirmaran el diagnostico clínico.
Algunos pacientes podrían alcanzar una sobrevida larga, en el caso del síndrome velocardiofacial, y los equipos multidisciplinarios deberán estar preparados para ofrecer un soporte apropiado en las diferentes etapas de la vida, y los riesgos de recurrencia y la asesoría genética deberían optimizarse con la disponibilidad de tales pruebas en nuestro medio.
Agradecimientos: Dra. Olga Soliz, Jefa de Laboratorio HMI, Dr. Abel Moncada, Inmunólogo HMI, Dra. Sandra Siacar, Endocrinóloga HMI, Dr. Erick Arzabe, Cirujano Maxilo Facial Hospital de Niño
1. Edelman L, et.al. A common molecular basis for rearrangement disorders on chromosome 22q11. Human Molecular Genetics 1999; 7:1157-67 [ Links ]
2 Frutos C. Estudio de la microdeleción cromosómica en 22q11 en neonatos con cardiopatías conotruncales y del arco aórtico. RELAN 1998;1:69-73 [ Links ]
3. Hokanson J. 22q11.2 microdeletions in adults with familial tetralogy of Fallot. Genetics in Medicine 2001:3:61–4.
4. Lindsay E. Chromosomal microdeletions: dissecting the 22q11 syndrome. Nature Reviews Genetics 2001;Nov.(2):858-68 [ Links ]
5. Scambler P. The 22q11 deletion syndromes. Human Molecular Genetics. 2000;16: 2421-6 [ Links ]
6. Marino B. Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet Med. 2001; 3:45-8. [ Links ]
7. Momma K. Tetralogy of Fallot associated with chromosome 22q11.2 deletion in adolescents and young adults. Genetics in Medicine 2001; 3:56–60.
8. Cuneo B. 22q11.2 deletion syndrome: DiGeorge, velocardiofacial, and conotruncal anomaly face syndromes. Current Opinion in Pediatrics 2001; 13:465–72
9. Swillen A, Devriendt K, Legius E, Fryns JP. Intelligence and psychosocial adjustment in velocardiofacial syndrome: a study of 37 children and adolescents with VCFS. J Med Genet 1997; 34:453–8.
10. Britt Ravnan J, Chen E, Golabi M, Lebo RV. Chromosome 22q11.2 microdeletions in velocardiofacial syndrome patients with widely variable manifestations. Am J Med Genet 1996; 66:250-6. [ Links ]
11. Driscoll DA, Salvin J, Sellinger B, et al. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet 1993; 30:813-7. [ Links ]
12. Scambler PJ, Kelly D, Lindsay E, et al. Velo-cardio-facial syndrome associated with chromosome 22 deletions encompassing the DiGeorge locus. Lancet 1992; 1:1138-9. [ Links ]

