










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
versión On-line ISSN 1024-0675
Rev. bol. ped. v.45 n.2 La Paz abr. 2006
CASO CLINICO
Craniopharyngioma, a case report
Drs.: Ariel Salas Mallea*, N. Claret Burgoa Medrano*, Corina Rocha Fernández*
* Médicos Residentes de Pediatría. Hospital del Niño “Dr. Ovidio Aliaga Uría”. La Paz-Bolivia. Correo electrónico: dr.ariel.salas@gmail.com
Resumen
Los craneofaringiomas representan aproximadamente 610% de los tumores cerebrales en la población pediátrica. Su manifestación clínica puede variar desde un estado asintomático hasta un amplio espectro de síntomas neurológicos, psicológicos, visuales y endocrinos. Presentamos el caso de un preescolar masculino de 2 años y 11 meses de edad con craneofaringioma gigante diagnosticado por tomografía simple de cerebro. Fue referido y hospitalizado por presentar desnutrición severa marasmática.
Rev Soc Bol Ped 2006; 45 (2): 98-101: tumores cerebrales pediátricos, craneofaringioma, desnutrición severa
Abstract
Craniopharyngioma represents approximately 6-10% of pediatric brain tumors. The clinical presentation of pediatric patients with craniopharyngioma consists of a broad spectrum of symptoms that range from asymptomatic to endocrine, visual, or psychological disorders. We describe a case of giant craniopharyngioma diagnosed by CT scan in a 35 months old child. He was referred and admitted to the hospital due to severe malnutrition.
Key Words
Rev Soc Bol Ped 2006; 45 (2): 98-101: pediatric brain tumors, craniopharyngioma, severe malnutrition.
Los tumores cerebrales son, después de las leucemias, los procesos malignos más frecuentes en la infancia y pueden presentarse a cualquier edad. El craneofaringioma representa aproximadamente 6-10% de los tumores cerebrales en la población pediátrica y su manifestación clínica puede ser muy variable1,2.
Preescolar masculino de 2 años y 11 meses de edad hospitalizado como desnutrido severo marasmático. Al momento de ingreso la madre refería un padecimiento de dos meses de evolución caracterizado por vómitos post-prandiales persistentes, hiporexia, disminución subjetiva y progresiva de peso además de compromiso del estado general. Es producto de primer embarazo, nacido de término por parto institucional sin complicaciones de una madre de 24 años sin antecedentes patológicos de importancia, con llanto inmediato al nacer y desarrollo psicomotor adecuado hasta el año de edad, a partir del cual se percibió falta de desarrollo del lenguaje receptivo y expresivo además de retraso del desarrollo motor caracterizado por imposibilidad de ponerse de pie y caminar sin apoyo.
Al examen físico de ingreso se encontraba en mal estado general y nutricional. El indicador peso para la talla era menor a -3 desvíos estándar (DE), existía emaciación visible y palidez mucocutánea generalizada. Los signos vitales estaban en parámetros normales. Neurológicamente llamaba la atención una respuesta verbal inapropiada, imposibilidad de bipedestación y aparente afectación visual manifestada por imposibilidad del paciente de seguir objetos con la mirada. Su perímetro cefálico fue de 48 cm. (adecuado para la edad) y las pupilas eran isocóricas reactivas al estímulo luminoso. El resto de la exploración física no mostró hallazgos patológicos.
Los exámenes de laboratorio solicitados (biometría hemática, proteínas, electrólitos y glucemia) se encontraban en parámetros normales.
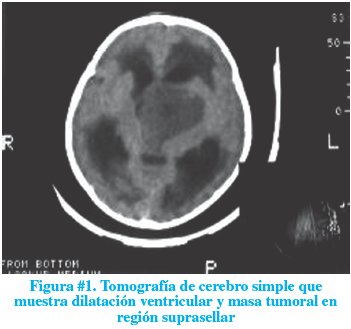
A las 48 horas de ingreso, presentó deterioro de su estado neurológico. Se tornó soporoso con puntuación Glasgow 9/15, reactivo sólo al estímulo doloroso y presentó signos clínicos de hipertensión endocraneana (vómitos explosivos, edema de papila y bradicardia para la edad). Se realizó una tomografía computarizada (TC) de cerebro simple que mostró un tercer ventrículo y ventrículos laterales con importante dilatación. A nivel de región supratentorial se observaba imagen atenuada con diámetros mayores de 53 x 50 mm sugerente de masa tumoral en región suprasellar que condicionaba severa hidrocefalia supratentorial (figura # 1).
Para el manejo de hidrocefalia e hipertensión endocraneana secundaria, se procedió a la colocación de una válvula de derivación ventrículo-peritoneal y se tomó muestra de líquido cefalorraquídeo (LCR) en búsqueda de blastos. Recibió además tratamiento con dexametasona. El examen citoquímico de LCR solicitado informó ausencia de formas blásticas, normoglucorraquia y proteinorraquia normal. Luego de cinco días, se recuperaron parcialmente las funciones motoras y sensitivas.
A los 20 días de tratamiento, presentó como complicación una ventriculitis, manifestada por respuesta inflamatoria sistémica, LCR compatible con proceso inflamatorio, presencia de reactantes de fase aguda elevados (volumen de eritrosedimentación y proteína C reactiva) además de nuevo deterioro neurológico. Se aisló S. aureus meticilino-sensible en hemocultivo y se inició tratamiento con vancomicina por 14 días.
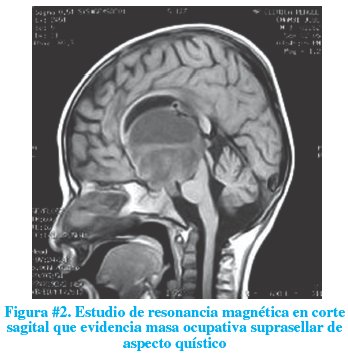
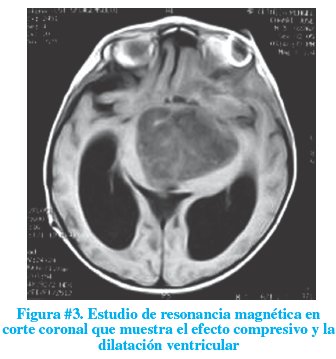
Luego de un mes de internación, se realizó un estudio de resonancia magnética el mismo que informó lesión ocupativa suprasellar de aspecto quístico e hidrocefalia no comunicante del sistema ventricular lateral (figuras # 2 y 3). Conocido este resultado, se solicitó un perfil endocrino el mismo que demostró normalidad.
En una segunda intervención quirúrgica, una vez estabilizada la condición clínica del paciente, se realizó biopsia del tumor para estudio histopatológico. El informe histopatológico confirmó el diagnóstico de craneofaringioma.
Actualmente el paciente se encuentra estable con amaurosis en espera de la realización de una intervención quirúrgica para la exéresis del tumor. El abordaje quirúrgico planeado para este paciente es una resección limitada seguida de radioterapia.
El craneofaringioma es un tumor de origen epitelial de la región sellar que se forma a partir de los restos embrionarios de la bolsa de Rathke. El tumor puede limitarse a la silla turca, o bien puede extenderse a través del diafragma sellar y comprimir la vía óptica, la protuberancia o el tercer ventrículo, produciendo hidrocefalia1,2. El paciente presentó esta última secuencia descrita.
Los craneofaringiomas representan entre el 5 y el 10% de todos los tumores intracraneales que se presentan durante la niñez. Tiene un leve predominio en el sexo masculino2,3. A pesar de su naturaleza benigna, el craneofaringioma puede causar daños permanentes y severos en el área visual, hipotalámica, endocrina y funciones neurocognoscitivas en general2. Como en la mayoría de los tumores intracraneales del niño, los signos y síntomas de hipertensión endocraneal son la primera manifestación de la enfermedad. La exploración clínica generalmente revela la existencia de alteraciones importantes en áreas tan elocuentes como la visual y la endocrina. La hipertensión endocraneana y la afectación visual están presentes en más del 50% de los casos al momento de la evaluación inicial. A pesar de la presencia de hidrocefalia, la macrocefalia es poco frecuente. La afectación endocrina se manifiesta por retraso en el crecimiento2,3.
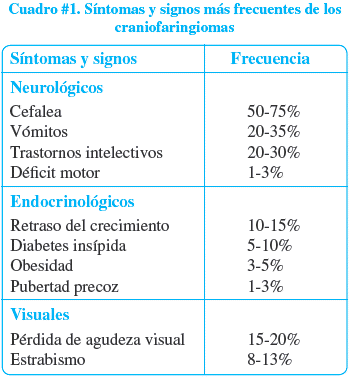
El cuadro clínico varía según la edad, en general, se dice que la hipertensión es el cuadro dominante en los lactantes, la pérdida de visión es el primer síntoma advertido entre los tres y los diez años y los problemas endocrinos pueden dominar el cuadro en la pubertad2. Los signos y síntomas más frecuentemente referidos como iniciales son los que se enumeran en el cuadro # 1.
Como describe la literatura, las manifestaciones clínicas en el paciente incluyeron desde un inicio síntomas de hipertensión endocraneana, compromiso visual y retraso del desarrollo psicomotor y de crecimiento. La macrocefalia estuvo ausente así como el compromiso endocrino.
Para el diagnóstico de esta entidad, se requieren estudios de imagen y pruebas de laboratorio. Desde el punto de vista imagenológico, el craniofaringioma es esencialmente un tumor que muchas veces es quístico y menos veces sólido y a veces puede ser mixto, con presencia de calcificaciones y asentado en la región suprasellar. La apariencia del craneofaringioma es característica en un estudio de TC. El modelo básico, aplicable en el 90% de los casos, sería el de un tumor localizado en la región suprasellar con un componente quístico, áreas de calcificación y zonas de captación de contraste yodado. La resonancia magnética es útil e imprescindible a la hora de planear la estrategia quirúrgica.
El diagnóstico histopatológico es también necesario en adición a estudios imagenológicos como la TC y la RM. Para el diagnóstico diferencial, se debe considerar todos los tumores supratentoriales1-4. En el paciente pudieron realizarse todos los estudios de imagen y laboratorio requeridos.
Con respecto al tratamiento, existen en la actualidad dos protocolos de tratamiento quirúrgico aprobados que consisten en la resección primaria total del tumor y la resección limitada o parcial seguida de radioterapia que resulta ser muy útil en la evolución del craneofaringioma4-7. Todavía están en estudio estrategias alternativas de tratamiento, especialmente para lactantes y niños de edad muy temprana que presentan tumores extensos, asociados con importante compromiso funcional. El empleo de técnicas innovadoras como la endoscopía para el diagnóstico y tratamiento, constituye la terapéutica quirúrgica menos agresiva, reduciendo el tiempo de recuperación y mejorando los resultados finales en dichos pacientes6. La resección total del tumor previene la recurrencia de quistes, sin embargo, la mayoría de estas lesiones se ubican adyacentes al tercer ventrículo y dificultan por lo tanto su abordaje. La colocación de un dispositivo ventrículo peritoneal bajo guía endoscópica evita la cirugía abierta. Bajo estas circunstancias también se ha demostrado que existen menos informes de revisión del dispositivo y posibles complicaciones que se encuentran relacionadas con su presencia6,7.
Referencias
1. Kuttesch JF, Ater JL. Tumores cerebrales en la infancia. En:Behrman RE, Kliegman RM, Jenson HB, eds. Tratado de Pediatría de Nelson. 17a ed. Madrid: Elsevier; 2004.p.1702-11. [ Links ]
2. Cohen BH. Garvin JH. Tumors of the central nervous system.In: Rudolph’s Pediatrics. 21st ed. 2003.p.955-78.
3. Duong M, Dinoulos JG, Gupta A, Bryk T, Saps M, Lorenzo C, et al. Index of suspicion. Pediatr Rev 2005; 26:23-33. [ Links ]
4. Maher CO, Raffel C. Neurosurgical treatment of brain tumors in children. Pediatr Clin N Am 2004;51:327-57. [ Links ]
5. Poretti A, Grotzer MA, Ribi K. Outcome of craniopharyngioma in children: Long-term complications and quality of life. Dev Med Child Neural 2004;46:220-9. [ Links ]
6. Kamikawa S, Inui A, Kobayashi N. Endoscopic treatment of hydrocephalus in children: a controlled study using newly developed Yamadori-type ventriculoscopes. Minim Invasive Neurosurg 2001;44:25-30 [ Links ]
7. Joki T, Oi S, Babapour B. Neuroendoscopic placement of Ommaya reservoir into a cystic craniopharyngioma. Childs Nerv Syst 2002;18:629-33. [ Links ]

