










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
versión On-line ISSN 1024-0675
Rev. bol. ped. v.44 n.2 La Paz jun. 2005
CASO CLINICO
Hiperplasia suprarrenal congénita por déficit de 21 hidroxilasa
Congenital adrenal hyperplasia due to 21 hydroxylase deficiency
Drs.: .Juan Pablo Hayes Dorado* Martha Eid de Pommier*, Walter Montero Justiniano*
*Medico pediatra, "Hospital Santa Cruz", Caja Petrolera de Salud, Santa Cruz de la Sierra. Bolivia
Articulo recibido 4/3/05, fue aprobado para publicación 10/7/05
El déficit de 21 hidroxilasa es la forma más frecuente de hiperplasia suprarrenal congénita ya que supone el 95% de los casos, La incidencia general de las formas clasicas es de aproximadamente 1/15.000 recién nacidos e implica la existencia de un hiperandrogenismo intraútero que condiciona la aparicion de un grado variable de virilización de los genitales externos en la mujer.
Describimos dos neonatos atendidos en el Hospital Santa Cruz de la Caja Petrolera de Salud por presentar genitales ambiguos. En ambos se diagnosticó hiperplasia suprarrenal congénita por déficit de 21 hidroxilasa, forma clásica y virilizante. El tratamiento instaurado fue en base a hidrocortisona por vía oral.
La ambiguedad de genitales en la etapa neonatal debe considerarse como una urgencia médica y se debe descartar hiperplasia suprarrenal congénita por que requiere un manejo inmediato.
Palabras Claves
Rev Soc Bol Ped 2005; 44(2)93-96: hiperplasia suprarrenal congénita, 21 hidroxilasa, genitales ambiguos.
The 21 e deficiency is the most frequent forrn of congenital adrenal hyperplasia because it means the 95% of the cases. The general incidence of the clasic form is nearly 1/15.000 newborn and implicates the existence of an fetal hyperandrogenism that influences the appearance of a variable grade of virilization of the external genitalia in the wornan.
We describe two newborn attended at the Hospital Santa Cruz of the Caja Petrolera de Salud by presenting arnbiguous genitalia. Both of them were diagnosed congenital adrenal hyperplasia due to 21 hydroxylase deficiency, clasic and virilizing. The treatment was hydrocortisone.
The ambiguous genitalia must consider as an medical urgency and congenital adrenal hyperplasia must be rule out because this disorder requires an inmediately treatment.
Key Words
Rev Soc Bol Ped 2005; 44(2)93-96: congenital adrenal hyperplasia, 21 hydroxylase, Ambiguous genitalia,
Introducción
La hiperplasia suprarrenal congénita engloba todos los trastornos hereditarios de la esteroidogenia suprarrenal de cortisol. El déficit de cortisol, que es el hecho común a todas ellas, produce por un mecanismo de retroalimentación negativa, un aumento de la producción de corticotropina (ACTH) y secundariamente una hiperestimulación e hipertrofia / hiperplasia del córtex adrenal motivando una elevación de los esteroides previos al bloqueo enzimático1.
En función del déficit enzimático se conocen cinco formas clínicas de hiperplasia suprarrenal congénita2, cuadro# 1.

Los estudios genéticos y clínicos han demostrado la existencia de formas graves y moderadas, en función del grado de afectación enzimática. En las formas graves o clásicas el déficit es completo e inician sus manifestaciones en la época fetal; en las formas moderadas o no clásicas el déficit es parcial y se manifiestan clínicamente en la infancia y la adolescencia, e incluso pueden pasar inad vertidas hasta la edad adulta.3
El déficit de 21 hidroxilasa (OH) es la forma más frecuente de hiperplasia suprarrenal congénita, ya que su pone el 95% de los casos .
El déficit de 21 OH presenta dos características fundamentales: insuficiencia suprarrenal e hiperandrogenismo, que derivan directa o indirectamente de la incapacidad de transformar 17 hidroxiprogesterona (17 OHP) en 11 desoxicortisol (déficit de secreción de cortisol) y progesterona en desoxicorticosterona (déficit de secreción de aldosterona), así como la acumulación de 17 OHP, androstenodiona, testosterona y de sus metabolitos respectivos2.
El espectro continuo de manifestaciones clínicas se clasifican en dos formas:
a) Clásicas (pérdida salina y virilizante simple).
b) No clásicas (sintomática y críptica).
La incidencia general de las formas clásicas es de aproximadamente 1/15 .000 y de las formas no clásicas de 1/1.000.1
Casos clínicos
Paciente N° 1: Recién nacido obtenido por cesárea, producto de primera gestación, 39 semanas de edad gestacional, 3540 g de peso, Apgar 9 -10, con genitales ambiguos (Prader IV) , ver figura #1.
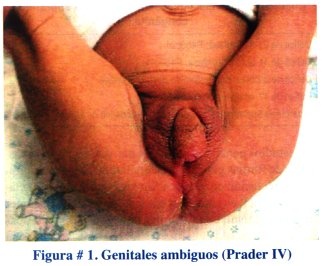
Se practicó una ecografía pélvica en la que se observó el útero y los anexos con características normales para la edad. El cariotipo reportó 46 XX. El nivel de 17 hidroxiprogesterona al 3er. día de vida fue de 95 ng/mL. Se diagnóstico hiperplasia suprarrenal congénita, forma clásica, virilizante y se inició tratamiento con hidrocortisona, 5 mg/día. Se controla al paciente mensualmente. Está programada la cirugía correctora de los genitales ambiguos a los 12 meses de edad.
Paciente N° 2: Recién nacido obtenido por parto normal , producto de primera gestación, 38 semanas de edad gestacional, 323 -1 g de peso, Apgar 8 -10, con genitales ambiguos (Prader 11), ver figura # 2.
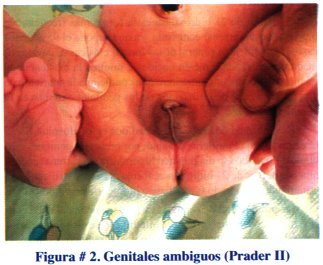
Se practicó unaecografía pélvica en la que se observó el útero y los anexos con características normales para la edad. El cariotipo reportó 46 XX. El nivel de 17 hidroxiprogesterona al 4to. día de vida fue de 87 ng/mL. Se diagnóstico hiperplasia suprarrenal congénita, forma clásica, virilizante y se inició tratamiento con hidrocortisona, 5 mg/día. Se controla al paciente mensualmente. Está programada la cirugía correctora de los genitales ambiguos a los 12 meses de edad.
Discusión
El diagnóstico hormonal del déficit de 21 OH se basa en la demostración de valores plasmáticos elevados de 17 OHP. En el déficit clásico de 21 OH, la 17 OHP basal se encuentra generalmente a las 48 horas de vida por encima de 20 ng/rnl, aunque en realidad se alcanzan valores superiores a 30-100 ng/ml.2
En las formas no clásicas el bloqueo es menos grave y la acumulación de 17 OHP puede ser muy variable, por lo que es aconsejable la realización de un test de ACTH en el que se demuestre la elevación de los valores de 17 OHP por encima de 10-15 ng/ml .
Todas las formas de hiperplasia suprarrenal congénita se heredan con carácter autosómico recesivo.3
El gen responsable del déficit de 21 hidroxilasa se denomina CYP21 B o CYP 21 y está localizado en el brazo corto del cromosoma 6p21.3, en la región III del sistema HLA. Todas las formas clínicas están asociadas a una anomalía en este gen, por lo que en la actualidad todos los pacientes deberían tener un diagnóstico genético, además del diagnóstico hormonal y clínico; paralelamente se debe hacer un estudio familiar que permita realizar el diagnóstico de portadores o de formas no clásicas oligosintomáticas y/o crípticas.4,5
A diferencia de otras enfermedades recesivas, donde es frecuente presentar la misma mutación en los dos alelos, en el déficit de 21 hidroxilasa los enfermos son frecuentemente heterocigotos compuestos o dobles heterocigotos: es decir tienen diferentes mutaciones génicas en cada alelo, una proveniente del padre y otra de la madre. Sólo en caso de mutaciones frecuentes o en el caso de consanguinidad se encuentran enfermos homocigotos para una determinada mutación. Los portadores o heterocigotos presentan un solo cromosoma mutado. y en principio no manifiestan signos clínicos. aunque sí una respuesta elevada de 17 OHP en el test de ACTH.3
La forma clásica de hiperplasia suprarrenal congénita por déficit de 21 hidroxilasa implica la existencia de un hiperandrogenismo intraútero que condiciona la aparición de macrogenitosomía en el varón y de un grado variable de virilización de los genitales externos en la mujer.2
En la forma pérdida salina. que es la expresión más grave de la enfermedad. existe un déficit tanto de cortisol como de aldosterona y se manifiesta en ambos sexos con crisis de pérdida salina en la época neonatal. Esta crisis de insuficiencia suprarrenal tiene una importante morbimortalidad si no se instaura un tratamiento adecuado.6
En la forma clásica virilizante simple, la síntesis de aldosterona no está tan gravemente alterada, por lo que se mantiene la homeostasis del sodio y no presentan crisis de pérdida salina. Las niñas son identificadas precozmente por la virilización de los genitales externos, pero los niños. y las niñas con una virilización leve suelen diagnosticarse tardíamente en la infancia cuando se hacen manifiestos los signos de hiperandrogenismo y la aparición de una seudopubertad precoz2.
En las formas no clásicas existe un hiperandrogenismo de aparición postnatal. Los síntomas más frecuentes en la infancia son pubarquia prematura, piel grasa con acné, aceleración del crecimiento y de la maduración ósea; en las niñas puede aparecer una moderada hipertrofia del clítoris. En la adolescencia y la edad adulta las mujeres pueden presentar irregularidades menstruales, hirsutismo, calvicie, ovario poliquístico, acné e infertilidad. Los varones afectados pueden presentar acné, oligospermia e infertilidad, pero la mayoría de las veces son asintomáticos.7
Las formas crípticas cursan únicamente con hallazgos hormonales, pero sin ninguna sintomatología, si bien actualmente se cree que pueden presentar eventualmente algún signo clínico de hiperandrogenisrno.7
Todos los pacientes con déficit clásico de 21 OH. así como los pacientes sintomáticos de las formas no clásicas deben tratarse con glucocorticoides, ya que así se suprime el exceso de secreción de hormona liberadora de corticotropina (CRH) y corticotropina (ACTH) y se reduce el exceso de esteroides sexuales de origen adrenal.8
La hidrocortisona es el tratamiento más fisiológico (tiene una potencia similar a la del cortisol endógeno, una corta vida biológica que minimiza el efecto sobre el crecimiento y sobre otros efectos adversos), la dosis es de 10 mg/m2/día, variable en función de la edad. Los neonatos son tratados habitualmente a una dosis de 5 mg/día. dividido en 3 dosis. que supone una dosis de 25 mg/m2/día, dosis suprafisiológica necesaria para suprimir los andrógenos adrenales y minimizar la posibilidad de desarrollar una insuficiencia suprarrenal. En los pacientes con formas no clásicas sintomáticas está indicado iniciar el tratamiento con glucocorticoides a dosis bajas, generalmente a mitad de dosis que en las formas clásicas.1,9
Los pacientes con pérdida salina requieren la administración de un mineralocorticoide, el más utilizado es el 9 alfa fluorhidrocortisona, 0.1 mg/día, dividido en 2 ó 3 dosis se requieren suplementos de cloruro de sodio (1-2 g/d), durante el primer año de vida.8
En situaciones de estrés o de enfermedad intercurrente, los pacientes requieren tratamiento hidroelectrolítico apropiado y aumentar la dosis de hidrocortisona, que debe administrarse por vía intravenosa.6
Está comprobado que mejora la talla final de los pacientes con hiperplasia suprarrenal al administrar hormona de crecimiento y un análogo de LHRH. 10
Respecto al tratamiento quirúrgico, la actitud terapéutica se inicia con la asignación precoz del sexo que debe ser la del sexo genético/gonadal, por la posibilidad de mantener la función reproductora. Se obtienen buenos resultados con la realización de la reconstrucción genital (clitoroplastía y vaginoplastía) en un mismo acto quirúrgico, hacia el segundo semestre de edad. El objetivo es la corrección completa de los genitales externos antes de los 18 meses de edad.
El buen control terapéutico durante la infancia y la adolescencia es fundamental para asegurar un crecimiento correcto, una maduración puberal normal y una ausencia de complicaciones a largo plazo: el objetivo es buscar la dosis mínima eficaz que garantice un buen crecimiento y una adecuada supresión de los andrógenos suprarrenales. Los parámetros de vigilancia incluyen datos clínicos como edad ósea, peso, talla y velocidad de crecimiento y hormonales. como determinación de 17 OHP, testosterona, delta 4 androstenodiona, ACTH y actividad de renina plasmática.7
En las gestaciones con riesgo de tener un hijo afecto de hiperplasia suprarrenal virilizante se ha conseguido frenar la producción de andrógenos suprarrenales fetales y disminuir la ambigüedad genital administrando dexametasona a la madre gestante; de esta manera se previene la virilización genital del feto mujer afectado. El tratamiento prenatal debe ir acompañado siempre de un adecuado diagnóstico genético prenatal.5,8
La detección neonatal sistemática, mediante la impregnación de sangre sobre papel secante entre el 3er y el 5to.día de vida, determinando las cifrasde 17OHP, permite apreciar la incidencia de la enfermedad, salvar a un cierto número de niños en particular del sexo masculino, que habrían muerto en ausencia de un diagnóstico correcto, e instaurar un tratamiento precoz, aunque no se llega a identificar la totalidad de recién nacidos con formas moderadas de la enfermedad. Se tomará en cuenta que los niveles de 17OHP presentan variaciones de acuerdo a la edad gestacional.11,12
Las alteraciones del desarrollo sexual se mani fiestan generalmente desde el nacimiento y requieren una investigación apropiada para determinar la causa de la anormalidad. establecer la terapia y decidir la asignación social del sexo del paciente. La ambigüedad de genitales en la etapa neonatal debe considerarse como una urgencia y se debe descartar hiperplasia sup rarrenal congénita. ya que esta entidad requiere manejo inmediato. El resto de las entidades no representan riesgo inmediato de morbilidad, pero es importante realizar la asignación sexual y la corrección quirúrgica lo más pronto posible, ya que cuando se realizan después de los dos años de edad, frecuentemente ocurren trastornos psicológicos y sociales en el paciente y en su núcleo familiar.13
El abordaje diagnóstico y terapéutico apropiado requiere una valoración particular y un diseño específico del plan de estudios de acuerdo a la edad y características del paciente se debe contar con un equipo multidisciplinario, integrado por especialistas en cirugía, urología, endocrinología, genética, ginecología, patología, pediatría, sicología, radiología y trabajo social. Se deben realizar determinaciones hormonales de sus precursores y receptores e incluso pruebas especializadas de genética molecular.4
El médico debe utilizar términos neutros, desde el primer contacto con el paciente; describirlo a los padres como "su bebé" y no como "el niño o la niña" o "su hijo o su hija"; hablar de "sus genitales y sus gónadas", evitando los términos "pene, clítoris, vagina, testículos y ovarios". Se evitará la asignación legal (registro civil) y religiosa (bautismo) del paciente hasta que se hay concluido el estudio y definido el sexo de asignación."
Referencia:
l. Merke D, Bornstein S. Congenital adrenal hyperplasia. Lancet. 2005;365(9477):2125-36.
2. New MI.An Update of Congenitul Adrenal Hyperplasia. Ann NY Acad Sci 2004;1038:14-43. [ Links ]
3. Trakakis E. 21-Hydroxylase deficiency: from molecular generics to clinical presentation. J Endocrinol Invest 2005;28(2):187-92 [ Links ]
4. Kosel S. Rapid second-tier molecular genetic analysis for congenital adrenal hyperplasia atrributable to steroid 21 hydroxylase deficiency. Clin Chem 2005:51(2):298-304. [ Links ]
5. Vakili R. Molecular analysis of the CYP21 gene and prenatal dignosis in families with 21-hydroxylase deficiency. Horm Res 2005;63(3):119-24 . [ Links ]
6. Perry R. Primary adrenal insufficiency in children. J Clin Endocrinol Metab 2005;90(6):3243-50. [ Links ]
7. Otten BJ. Puberty and fertility in congenital adrenal hyperplasia. Endocr Dev 2005;8:54-66. [ Links ]
8. Warne G. Hormonal therapies for individuals with intersex conditions:protocol for use. Treat Endocrinol 2005;4(1):19-29. [ Links ]
9. Khadilkar V. lrnpact of availability of oral hydrocortisone on growth of children with CAH. Indian J Pediatr 2005;72(4):19-29. [ Links ]
10. Lin-Su K. Treatment with growth horrnone and luteinizing hormone releasing hormone analog irnproves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2005;90(6):3318-25. [ Links ]
11. Votava F. Estimation of the false-negativa rate in newborn screening for congenital adrenal hyperplasia. Eur J Endocrinol 2005;152(6):869-74. [ Links ]
12. Brown J, Warne G. Practical managemem of the intersex infant. J Pediatr Endocrinol Metab 2005;18(1):3-23. [ Links ]

