










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
versión On-line ISSN 1024-0675
Rev. bol. ped. v.43 n.3 La Paz ago. 2004
CASO CLINICO
Tumores adrenocorticales: a propósito de dos casos
Drs.: Germán Quevedo P.*, Juan Pablo Hayes D.**, Rony Colanzi Z.***, Catherine Arispe****
* Cirujano - Urólogo Pediatra. Jefe del Servicio de Cirugía Pediátrica. Hospital Universitario Japonés. Santa Cruz de la Sierra, Bolivia.
** Médico pediatra. Hospital Santa Cruz. Caja Petrolera de Salud. Santa Cruz de la Sierra, Bolivia.
*** Médico patólogo. Hospital Universitario Japonés. Santa Cruz de la Sierra, Bolivia.
**** Residente II del Servicio de Pediatría. Hospital Universitario Japonés. Santa Cruz de la Sierra, Bolivia.
Artículo recibido 30/10/2004, fue aprobado para publicar 8/12/2004.
Resumen
Los tumores adrenocorticales son poco frecuentes en la población infantil. En esta comunicación se describen dos pacientes atendidos en el Servicio de Cirugía Pediátrica del Hospital Universitario Japonés, de la ciudad de Santa Cruz de la Sierra, Bolivia, presentando precozmente vello pubiano. La tomografía abdominal contrastada, reveló en ambos casos, tumor adrenocortical y el tratamiento efectuado fue la extirpación de la masa tumoral. Se confirmó el diagnóstico por Anatomía patológica concluyendo tratarse de un carcinoma adrenocortical y de otro adenoma adrenocortical.
Palabras Claves:
Rev Soc Bol Ped 2004; 43 (3): 159-63: tumor adrenocortical, carcinoma adrenocortical, adenoma adrenocortical.
Abstract
The adrenocortical tumors are not very frequent in the infantile population. We describe two cases attended in the ward of Pediatric Surgery of the Hospital Universitario Japonés, at Santa Cruz de la Sierra, Bolivia.
The patients, a three years-old girl and a four years-old boy had a precocious pubic body hair. The CT revealed in both cases adrenocortical tumors, and they were treated with the extirpation of the tumoral mass. Their diagnosis were confirmed by pathological reports: an adrenocortical carcinoma in the girl and an adrenocortical adenoma in the boy.
Key words:
Rev Soc Bol Ped 2004; 43 (3): 159-63: Adrenocortical, adrenocortical carcinoma, adrenocortical adenoma.
Introducción
Los tumores adrenocorticales son poco frecuentes; afectan solamente una a dos personas de cada millón; la incidencia estimada es de 1 por 1,7 millones de personas.(1) En la población infantil son raros; en 1865 se reportó el primer caso en un niño de tres años de edad. En el Registro Internacional de Tumores Adrenocorticales Pediátricos se han documentado 254 casos hasta marzo del 2004. En los Estados Unidos de Norteamérica, la incidencia de cáncer en menores de 20 años de edad es de 10.000 nuevos casos por año; de éstos, del 2 al 12% son carcinomas, entre los cuales sólo del 3 al 6% son carcinomas adrenocorticales (reportándose sólo de19 a 25 casos nuevos por año). En el Sur de Brasil es 10 veces más frecuente, presentando el 65 % de los pacientes pediátricos con esta patología una edad inferior a los 5 años.(2)
Casos clínicos
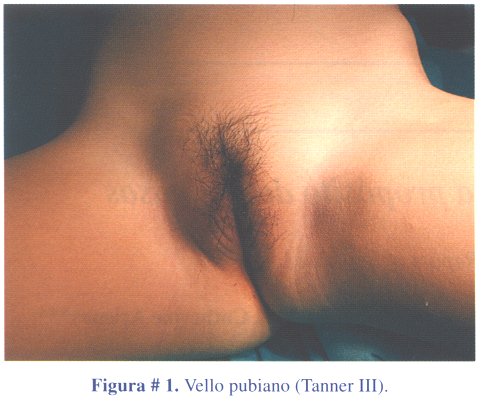
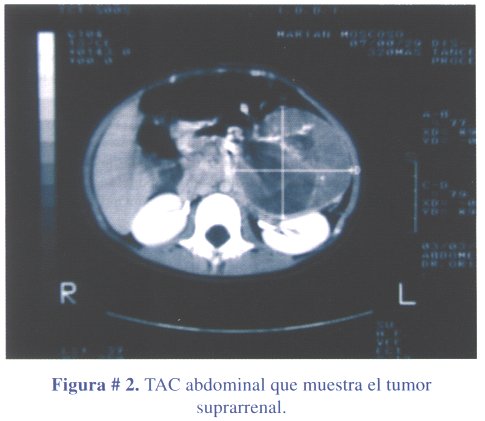
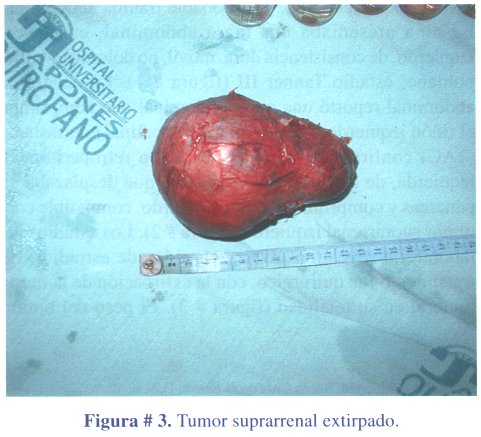
1º caso clínico: niña de 3 años y 6 meses de edad, que acudió a consulta médica por presentar dolor abdominal y vómitos a repetición, por probable trauma abdominal. La niña presentaba una masa abdominal, en flanco izquierdo, de consistencia dura, móvil, no dolorosa y vello pubiano, estadio Tanner III (figura # 1). La ecografía abdominal reportó una masa abdominal que comprimía el riñón izquierdo. La tomografía abdominal contrastada (TAC) confirmó la masa de ubicación retroperitoneal izquierda, de densidad heterogénea y que desplazaba el páncreas y comprimía el riñón izquierdo, compatible con tumor suprarrenal izquierdo (figura # 2). Los estudios de laboratorio revelaron niveles elevados de estradiol. El tratamiento fue quirúrgico, con la extirpación de la masa tumoral en su totalidad (figura # 3). El peso del tumor fue de 300 gramos.
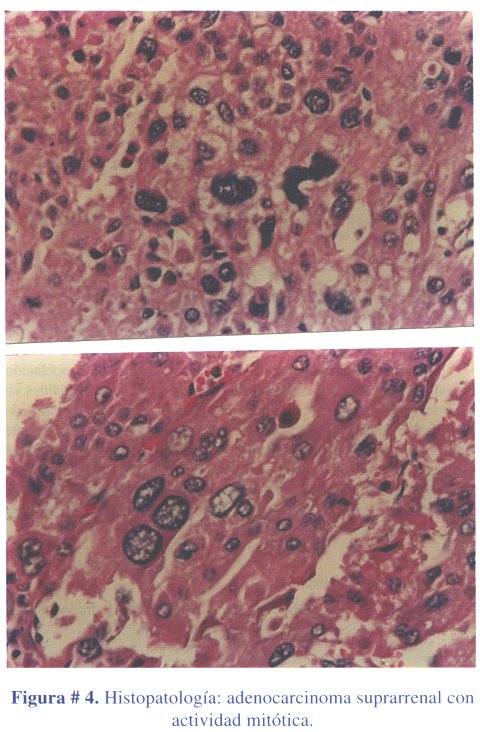
El informe anatomopatológico confirmó tratarse de un adenocarcinoma suprarrenal (figura # 4).
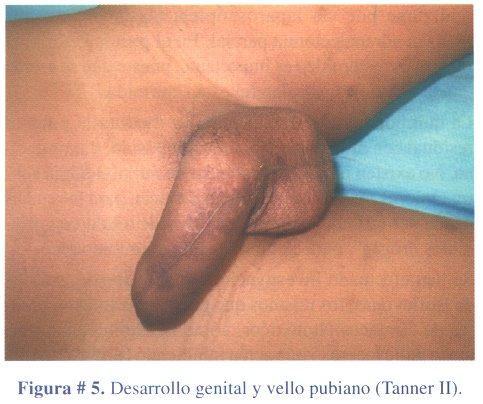
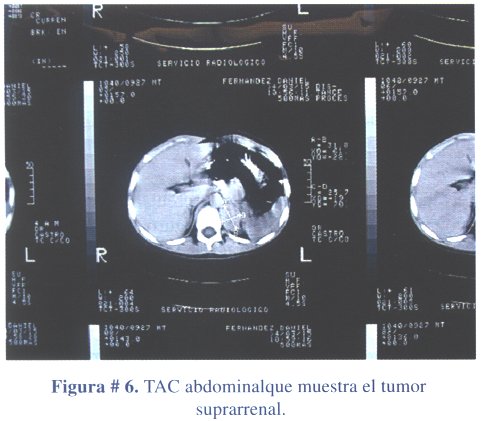
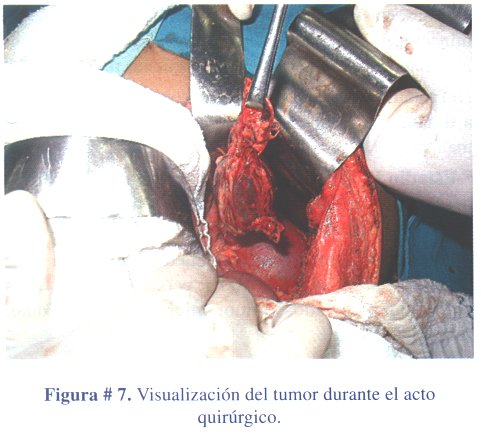
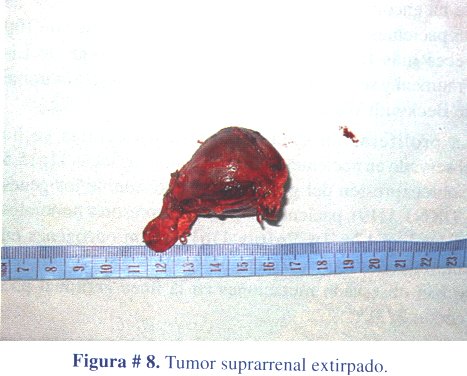
2º caso clínico: niño de 4 años de edad, que acudió a consulta por presentar caracteres sexuales secundarios: vello pubiano y crecimiento escrotal, testicular y peniano (figura # 5). La ecografía abdominal reveló una masa ocupante en glándula suprarrenal izquierda, hallazgo confirmado con una TAC abdominal contrastada (figura # 6). Los estudios de laboratorio mostraron niveles elevados de sulfato de dehidroepiandrosterona. El tratamiento fue quirúrgico (figura # 7) extirpando el tumor en su totalidad (figura # 8). El informe anatomopatológico concluyó tratarse de un adenoma de corteza suprarrenal izquierda.
Discusión
Los tumores adrenocorticales generalmente se presentan en menores de cinco años de edad; la relación sexo femenino / masculino es de 1.6 : 1. Los síndromes clínicos endócrinos que pueden observarse en los pacientes son:
síndrome de Cushing, virilización, formas mixtas, feminización y síndrome de Conn (hiperaldosteronismo); el 10% de los tumores son no funcionales.(3)
La virilización se presenta en el 80% de los pacientes (vello pubiano, facial, cambio del tono de la voz, acné, hirsutismo e hipertrofia de clítoris en las niñas; pubertad precoz periférica en los niños), el síndrome de Cushing (cara de "luna llena", ganancia ponderal, distribución típica del tejido graso, hipertensión arterial y estrías) se acompaña de virilización en el 30% de los casos; la feminización se observa sólo en el 2.2% de los pacientes (ginecomastia) y el síndrome de Conn representa el 1.6 % de los casos (pacientes con cefalea, debilidad muscular proximal, poliuria y taquicardia).(4)
Los signos y síntomas más frecuentemente observados son: vello pubiano en el 91% de los casos, acné en el 72%, hipertrofia de clítoris en el 62%, cambio del tono de la voz e hipertensión arterial en el 55% y vello facial en el 50%; el tumor es palpable en el 48% de los pacientes.(5)
Se ha encontrado predisposición genética en el 50% de los pacientes con tumores adrenocorticales; éstos son 100 veces más frecuentes en niños con síndrome de Li-Fraumeni y se los ha relacionado también con el síndrome de Beckwith Wiedemann.
La proliferación monoclonal de las células se ha observado en pacientes con duplicación del locus 11p15.5 (sobreexpresión del gen IGF2 y supresión de los genes CDKN1, H19), pacientes con genes supresores tumorales en los loci 17p, 1p, 2p16 y 11q13 y con oncogenes en determinados loci de los cromosomas 4, 5 y 12; también se han observado mutaciones en la línea germinal p53 (codon 273).(6-7)
Se ha descrito que el peso medio de los tumores extirpados es de 126 g (rango de 2 a 6.000 g) y que la incidencia es similar en la glándula adrenal izquierda y en la derecha representando los tumores bilaterales el 1.3% de los casos, siendo raros los tumores ectópicos.(8)
Las etapas tumorales se pueden definir mediante la clasificación de TNM, ver cuadro #1. Respecto a la clasificación patológica del tumor adrenocortical en pacientes pediátricos es dificultosa, incluso para patólogos experimentados y se usa el sistema de Weiss o modificado por Bugg.(9)

Se ha efectuado el diagnóstico desde tres días hasta cinco años del inicio de manifestaciones clínicas, por lo que se ha sugerido que todo menor de cuatro años con vello pubiano debe considerarse con carcinoma adrenal, hasta demostrarse lo contrario.
El diagnóstico por imágenes se efectúa mediante la ultrasonografía (80 a 90% de sensibilidad), tomografía axial computarizada (98 a 99%) o resonancia nuclear magnética (sensibilidad del 100%). La tomografía por emisión de positrones con F-18 fluor-desoxi-glucosa (FDG PET) identifica mejor las lesiones malignas que la PET con 11 C metomidate (MTO).(10 -12)
Mediante pruebas de laboratorio se evalúan los niveles de sulfato de dehidroepiandrosterona, delta 4 androstenediona, testosterona, cortisol urinario, 17 hidroxiprogesterona, actividad de renina plasmática y aldosterona.(13)
Los niveles elevados de cortisol, testosterona, estradiol y sustratos intermediarios, obtenidos en sangre de la vena suprarrenal, durante la cirugía, han confirmado que estas hormonas son directamente liberadas desde el tumor y no resultantes de la conversión periférica. Por otro lado, se ha comprobado que los niveles de factor de crecimiento endotelial vascular (VEGF) en pacientes con tumores adrenales (especialmente carcinomas) son significativamente mayores que los observados en personas sanas.(14)
El tratamiento es quirúgico y consiste en la extirpación de la masa tumoral; la adrenalectomía total puede realizarse por vía laparoscópica; algunos cirujanos realizan adrenalectomía parcial. En el postoperatorio, el reemplazo esteroideo es importante, puesto que se asume que la glándula contralateral está suprimida.(15-20)
La quimioterapia se indica para las metástasis y tumor residual con mitotano, cisplatino, etopósido, doxorubicina. No existen protocolos estándar de quimioterapia contra tumores malignos con baja incidencia, para los cuales puede ser útil el "análisis de cultivo de tejido con diferentes drogas", para elegir el agente quimioterápico.(21-22)
Se han efectuado investigaciones con carboxiderivados de isoflavonas, los mismos que pueden ser transportadores de fármacos citotóxicos, como la daunomicina (fármaco anticanceroso que actúa inhibiendo la transcripción del DNA), siendo el blanco del conjugado "daunomicina-carboxiderivado", las células H295R del carcinoma adrenocortical, con receptores estrogénicos ("quimioterapia dirigida"). La radioterapia es de uso excepcional en niños.(21,23)
Se ha realizado, como medida terapéutica, la inyección percutánea de etanol, en metástasis hepáticas de carcinoma adrenocortical, en una niña de dos años de edad, con evolución favorable (seguimiento clínico de 3 años).(24)
La ablación por radiofrecuencia percutánea, dirigida por ecografía, es un procedimiento seguro y bien tolerado, potencialmente útil para el tratamiento de carcinomas no resecables o metástasis, siendo más efectiva en tumores de 5 cm o más pequeños.(25)
Se ha observado que el tiempo de recurrencia varía de uno a 48 meses, después de la cirugía (media de seis meses) y que las metástasis generalmente se presentan en hígado, pulmones y nódulos linfáticos, aunque se han descrito incluso en la tiroides. Excepcionalmente el carcinoma adrenocortical en los niños puede recurrir años después, en sitios poco comunes, en los cuales las determinaciones hormonales pueden representar indicadores insuficientes de la existencia de pequeñas metástasis.(26,27)
Del estadio del tumor depende la sobrevida del paciente a 6 años del diagnóstico y se clasifica de la siguiente manera:
* Estadio I.- Resección completa; márgenes negativos; peso menor de 200g; normalización de niveles hormonales; ausencia de metástasis: sobrevida mayor al 90%.
* Estadio II.- Resección completa del tumor; márgenes negativos; peso mayor de 200 g; persistencia de niveles hormonales elevados; tumor microscópico residual: sobrevida del 52%.
* Estadio III.- Resección incompleta; tumor inoperable: sobrevida del 25 %.
* Estadio IV.- Metástasis a distancia: sobrevida del 10%.
La sobrevida, aplicando el sistema de Weiss, es la siguiente a 5 años del diagnóstico: Comportamiento benigno.- Puntaje de Weiss menor o igual a 3: Sobrevida del 100%; comportamiento maligno.- Puntaje de Weiss mayor a 3: Sobrevida del 62%.
En el sistema de Weiss se analizan nueve parámetros: grado nuclear, rango mitótico, mitosis atípica, caracteres del citoplasma, arquitectura de las células tumorales, necrosis, invasión de estructuras venosas, sinusoidales e invasión de la cápsula del tumor.(19)
En general, se ha observado que son indicadores de peor pronóstico: El mayor tamaño del tumor, la resección parcial del mismo y presencia de metástasis al momento del diagnóstico.(14)
Referencias
1. Tupikowski W. Adrenocortical carcinoma and its treatment. Postepy Hig Med 2004;58:27-36. [ Links ]
2. Ghazi A et al. Cortisol and estradiol secretion by a virilizing adrenocortical tumor in a prepubertal girl. J Pediatr Endocrinol Metab 2004; 17: 235-8. [ Links ]
3. Michalkiewicz E. Clinical and outcome characteristics of children with adrenocortical tumors. J Clin Oncol 2004;22:838-45. [ Links ]
4. Narasimhan K. Adrenocortical tumors in childhood. Pediatr Surg Int 2003;19:432-5. [ Links ]
5. Cordera F. Androgen secreting adrenal tumors. Surgery 2003;134:874-80. [ Links ]
6. Sidhu S. Clinical and molecular aspects of adrenocortical tumourigenesis. J Surg 2003;73:727-38. [ Links ]
7. Hara F et al. A child with adrenocortical carcinoma who underwent percutaneous ethanol injection therapy for liver metastasis. J Pediatr Surg 2003; 38 :1237-40. [ Links ]
8. Ribeiro R. Adrenocortical tumors in children. Braz J Med Biol Res 2000;10:1225-34. [ Links ]
9. Bonfig W. Virilising adrenocortical tumours in children. Eur J Pediatr 2003;162:623-8. [ Links ]
10. Ihara M. Diagnosis and treatment for adrenocortical carcinoma. Gan To Kagaku Ryoho 2004;31:342-5. [ Links ]
11. Minn H. Imaging of adrenal incidentalomas with PET using (11) C-metomidate and (18) F-FDG. J Nucl Med 2004;45:972-9. [ Links ]
12. Zettinig G. Positron emission tomography imaging of adrenal masses: (18) F-fluorodeoxyglucose and the 11beta-hydroxylase tracer (11)C-metomidate. Eur J Nucl Med Mol Imaging 2004;31:1224-30. [ Links ]
13. Walz M. Partial versus Total Adrenalectomy by the Posterior Retroperitoneoscopic approach: Early and Long-term Results of 325 Consecutive Procedures in Primary Adrenal Neoplasias. World J Surg 2004; 29:524-9. [ Links ]
14. Zacharieva S. Circulating vascular endothelial growth factor and active renin concentrations and prostaglandin E2 urinary excretion in patients with adrenal tumors. Eur J Endocrinol 2004;150:345-9. [ Links ]
15. Stewart J. A surgical approach to adrenocortical tumors in children: the mainstay of treatment. J Pediatr Surg 2004;39:759-63. [ Links ]
16. Meyer A. Experience with the surgical treatment of adrenal cortical carcinoma. Eur J Surg Oncol 2004;30:444-9. [ Links ]
17. Kageyama Y. Portless endoscopic adrenalectomy via a single minimal incision using a retroperitoneal approach: experience with initial 30 cases. Int J Urol 2004;11:693-9. [ Links ]
18. Porpiglia F. Is laparoscopic adrenalectomy feasible for adrenocortical carcinoma or metastasis? BJU Int 2004;94:1026-9. [ Links ]
19. Lucon A, Pereira M, Mendoca B. Adrenocortical tumors. Rev Hosp Clin Fac Med S Paulo 2002;57:251-6. [ Links ]
20. Sturgeon C. Laparoscopic adrenalectomy for malignancy. Surg Clin North Am 2004; 84: 755-74. [ Links ]
21. Hovi L. Adrenocortical carcinoma in children: a role for etoposide and cisplatin adjuvant therapy. Med Pediatr Oncol 2003;40:324-6. [ Links ]
22. Yoshimasu T. Histoculture drug response assay guided concurrent chemoradiotherapy for metastasis from adrenocortical carcinoma. Gan To Kagaku Ryoho 2004;31:435-7. [ Links ]
23. Somjen D. Carboxy derivatives of isoflavones as affinity carriers for cytotoxic drug targeting in adrenocortical H295R carcinoma cells. J Endocrinol 2003;179:395-403. [ Links ]
24. Khayat C. Rhabdomyosarcoma, osteosarcoma, and adrenocortical carcinoma in a child with a germline p53 mutation. Pediatr Blood Cancer 2004;43:683-6. [ Links ]
25. Wood B. Radiofrequency ablation of adrenal tumors and adrenocortical carcinoma metastases. Cancer 2003;97:554-60. [ Links ]
26. Valo I. Thyroid metastases of an adrenocortical carcinoma 41 years after the diagnosis of the primary tumor. Ann Pathol 2004;24:264-7. [ Links ]
27. Jacobsen B. Late relapse of adrenocortical carcinoma in Beckwith-Wiedemann syndrome. Acta Pediatr 2003;92:439-43. [ Links ]

