










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
On-line version ISSN 1024-0675
Rev. bol. ped. vol.42 no.2 La Paz June 2003
CASO CLINICO
Esclerodermia o esclerosis sistémica. A propósito de un caso
Scleroderma
Drs. Jorge Barriga Oropeza,* Consuelo Murillo Sánchez*, Julio Agreda Guerrero**
* Médico pediatra. Hospital Daniel Bracamonte. Potosí - Bolivia
** Residente de pediatría. Hospital Daniel Bracamonte. Potosí - Bolivia
Resumen
Aprovechando el caso de una niña que se presentó en el Hospital Daniel Bracamonte, de la ciudad de Potosí, se describe la enfermedad que engloba a un grupo de enfermedades que pertenecen al grupo de las colagenosis. La esclerodermia (del griego esclero-dura y dermia-piel) en una enfermedad rara de naturaleza autoinmune y todas ellas tienen como síntomas comunes el endurecimiento de la piel y adelgazamiento cutáneo. La esclerosis sistémica no solo afecta al sistema autoinmune, sino también a la pared de los vasos sanguíneos y el tejido conectivo.
Palabras Claves
Rev Soc Bol Ped 2003; 42 (2): 97-9: colagenosis, esclerosis sistémica, esclerosis sistémica progresiva, esclerosis sistémica progresiva familiar.
Abstract
We described a girl with scleroderma that came to the Hospital Daniel Bracamonte, in Potosí, and it is included in the group of the colagenosis. This rare entity belongs to the autoimmune diseases. The main characteristics are hardening of the skin and thinner of the cutaneous layer. The systemic sclerosis affects not only the immune system but also the wall of the arterial vessels and connective tissue.
Key Words
Rev Soc Bol Ped 2003; 42 (2): 97-9: colagen disease, scleroderma, systemic scleroderma, systemic progressive scleroderma-systemic familiar scleroderma.
Caso Clínico
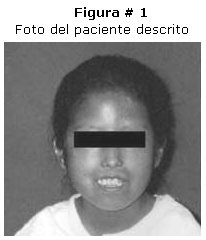
Paciente de sexo femenino de 12 años de edad, 22 kilos de peso que ingresa al hospital por presentar, deformidad de las articulaciones metacarpo falángicas, rigidez y cambios en la coloración de la piel, dolor articular en extremidades superiores, perdida de peso, evolución aproximada de dos años. Al examen físico se puede apreciar: cara con piel seca, disminución de la elasticidad ausencia de arrugas faciales. Extremidades superiores hipotroficas, hiporefléxicas, con perdida de la elasticidad de la piel e hiperpigmentación en toda su extensión. Manos y dedos con articulaciones interfalángicas en dorsiflexión, manchas hipocrómicas, rigidez en región de articulaciones interfalangicas y motilidad limitada. Figura # 1, # 2 y # 3.
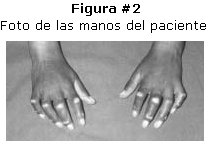
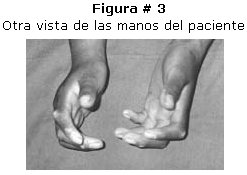
Los exámenes radiológicos demostraron osteoporosis, y los de laboratorio ASTO elevado, VSG aumentada, anemia leve y el resto de otros resultados sin particularidades. La biopsia fue compatible con el diagnóstico.
Discusión
Las esclerodermias son un grupo de enfermedades cuya causa se desconoce y que se caracterizan principalmente por una producción excesiva de colágeno que se acumula en ciertas partes de la piel produciendo un endurecimiento de la zona afectada, pudiendo además afectar a otros órganos, tales como el aparato digestivo, pulmón, riñón y corazón(1).
No se tiene datos exactos sobre su incidencia, especialmente en nuestro medio en la que se presentan casos muy raros y aislados, sin embargo en los Estados Unidos de América se registran aproximadamente trescientos mil personas con esta enfermedad, es más frecuente en mujeres que en hombres en una relación de 4:1. No se considera una enfermedad ni cancerosa ni contagiosa, no influyen factores hereditarios, solo existen factores inmunológicos y ambientales que podrían coadyuvar a que en determinado momento ciertos individuos desarrollen la enfermedad.
Se clasifica en: 1) Esclerosis localizada (morfea, en banda o lineal, en golpe de sable); 2) Esclerosis sistémica (esclerodermia sistémica limitada, esclerodemia sistémica difusa)(1-4).
Los tres fenómenos patogénicos de mayor importancia son: pérdida de la integridad vascular, pérdida del mecanismo regulador que normalmente controla el trauma y respuesta autoinmune(5-6).
1. Esclerodermias localizadas
Son más frecuentes en niños que en adultos, son más benignas, por cuanto no involucran otros órganos del cuerpo que no sea la piel.
a) Esclerodermia morfea. Se presenta hiperpigmentación de la piel en forma de placas o parches, la piel es clara, brillante endurecida y tensa.
b) Esclerodermia lineal o en banda. Aparece a lo largo de un miembro (pierna o brazo), causando atrofia de las masas musculares subyacentes, y puede ocasionar acortamiento del miembro afectado, si se desarrolla a través de una articulación puede provocar contractura o pérdida del movimiento.
c) Esclerodermia en golpe de sable. Afecta la piel de la región frontal y/o lateral de la cara.
2. Esclerosis sistémica progresiva
a) Esclerosis sistémica limitada. Se caracteriza por el hecho de que el fenómeno de Raynaud puede presentarse aisladamente muchos años antes de la piel. Este tipo de esclerosis sistémica tiene anticuerpos anticentrómero detectables.
b) Esclerosis sistémica difusa. Es la forma más grave se inicia dentro del primer año después de la afectación de la piel, la fibrosis pulmonar es precoz, existe también afectación del riñón se caracteriza con la llamada Crisis renal esclerodermica, la cual se asocia a cifras elevadas de tensión arterial, entre un 20 al 30 % de los pacientes con este tipo de esclerodermia tiene presente el anticuerpo anti-scl70(antitopoisomerasa I).
Las manifestaciones clínicas más importantes incluyen:
1. Afectación Cutánea: Evoluciona en tres fases.
a) Fase inicial o edematosa.- Afecta fundamentalmente a los dedos de las manos.
b) Fase intermedia o indurativa.- Aparece la piel engrosada y tirante, adquiere una textura dura con pérdida de elasticidad y desaparición de pliegues cutáneos, se acompaña de rigidez articular, así como de un rostro carente de expresividad con microstomia (disminución de la apertura bucal). La cara parece una máscara
c) Fase atrófica.- Se caracteriza por la presencia de atrofia (disminución de volumen y peso de un órgano) y adelgazamiento epidérmico.
2. Fenómeno de Raynaud: Es el más molesto de los síntomas de la enfermedad, es capaz de provocar ulceraciones muy dolorosas de las yemas de los dedos.
3. Síndrome de Sjögren: Consiste en la presencia de resequedad de las membranas mucosas especialmente en los ojos y boca principalmente.
4. Síndrome de C.R.E.S.T.: Se presenta en la variedad de esclerodermia sistémica limitada y se caracteriza por: Calcinosis, Fenómeno de Raynaud, Transtornos funcionales y estrechamieno del Esófago, Esclerosis de la piel y Telangiectaias.
5. Manifestaciones gastrointestinales: Esta enfermedad puede afectar prácticamente todo el tubo digestivo tanto en su estructura como en su forma, siendo el esófago el más comprometido, estas anormalidades causan en general intolerancia a los alimentos que en definitiva generan un marcado grado de desnutrición.
6. Fibrosis pulmonar: A causa de este compromiso se presenta disnea de esfuerzo, tos crónica, la fibrosis interfiere el intercambio gaseoso en los alvéolos pulmonares.
7. Manifestaciones músculo esqueléticas: La rigidez articular y las poliartralgias se presentan en la etapa inicial y a menudo representan la primera manifestación de la enfermedad. La afección tenosinovial se manifiesta por la presencia de frotes palpables y gruesos, semejantes a los que produce el cuero. La afección muscular puede manifestarse a través de la atrofia muscular por desuso o una miopatia inflamatoria caracterizada por la debilidad muscular y la elevación de las "enzimas de escape".
8. Manifestaciones renales: Son frecuentes también las alteraciones renales con la consecuente insuficiencia renal asociada a hipertensión arterial.
Los hallazgos radiológicos pueden incluir a nivel de los huesos: osteopenia, acroosteolisis, calcinosis. En el tórax: fibrosis intersticial pulmonar en casos avanzados puede observarse una imagen denominada "panal de abejas" en el esófago: dilatación del tercio inferior3.
Se consideran los siguientes criterios diagnósticos. Ver cuadro # 1.

Los principales hallazgos de laboratorio que nos permiten confirmar el diagnostico son: VSG elevada, anemia, hipergamma-globulinemia (IgG), FAN Positivo, presencia de diferentes anticuerpos específicos y biopsia característica de piel y de pulpa digital(5).
El esquema de tratamiento debe incluir medidas generales entre las que cabe destacar la educación del paciente y su familia, un programa apropiado de fisioterapia y rehabilitación, evitar traumatismos repetidos, evitar la exposición al frío, y mantenerse confortablemente abrigado con especial énfasis en las manos y los pies.
En cuanto al tratamiento de fondo las acciones terapéuticas son tanto locales: ultrasonido, masajes, aplicación de cremas humectantes para reblandecer la piel asociadas a vitamina E; como sistémicas: D.penicilamida, colchicina, metotrexato, prednisona, nifepidina, indobufeno, retinoides aromáticos, hidroxicloroquina, minocliclina. Dos nuevas drogas abren esperanzas en el tratamiento de esta enfermedad: rottlerin, que inhibe en un 90% la producción de colágeno y la relaxina que mejora la movilidad y disminuye el engrosamiento de la piel(6).
Referencias
1. Esclerodermia: un misterio que aún no se revela. Buena Salud . Bibliomed Inc. www.buenasalud.com 2001. [ Links ]
2. Esclerodermia. http://members.tripod.com/RENDILES/ESCLERO-DERMIA.html.2002 [ Links ]
3. Esclerodermias en la infancia. www.angelfire.com/ri/reuma/ED.html.2001 [ Links ]
4. Cuttica RJ, Garay SM. Sclerodermia. En: Meneghello JR, Fanta E, Paris E, Puga TF,eds. Pediatría. 5ta ed. Buenos Aires: Editorial Médica Panamericana; 1997.p.1195-200. [ Links ]
5. Miller ML. Esclerodermia. En: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson Tratado de Pediatría. 16 ed. Madrid: McGraw Hill Interamericana; 2000.p.789-92. [ Links ]
6. Seibold JR. et al. Safety and pharmacokinetics of recombinant human relaxin in systemic sclerosis. Scleroderma Program, University of Medicine and Dentristry of New Jerser-Robert Wood Johnson Medical School, New Brunswick O8903-0019. USA. 2002 www.ncbi.nlm.nih.gov. [ Links ]

