










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO Compartir

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
versión On-line ISSN 1024-0675
Rev. bol. ped. v.41 n.1 La Paz ene. 2002
ACTUALIZACIONES
Síndrome de dificultad respiratoria aguda
Acute respiratory distress syndrome
Dr.: Jorge Salazar*
*Médico Pediatra-Intensivista. Unidad de Terapia Intensiva. Hospital del Niño “Dr. Ovidio Aliaga Uría”. La Paz - Bolivia
Dirección: calle Mayor Zubieta sin número. Miraflores
Introducción
La lesión pulmonar en el paciente pediátrico puede deberse a distintas causas, que originan una respuesta endotelial muy exacerbada, produciéndose lesión pulmonar y según la gravedad de esta, se puede denominar daño pulmonar agudo (DPA) o síndrome de dificultad respiratoria aguda (SDRA). Esta entidad en el transcurso del tiempo fue tomando diferentes nombres y calificativos desde el pulmón de bomba, en pacientes post operados de corazón hasta el síndrome de insuficiencia respiratoria progresiva del adulto en el niño, sin embargo en el consenso americano europeo de ARDS en 1994 se eliminaron las anteriores denominaciones para poder tener una misma nomenclatura en el ámbito mundial.
La frecuente asociación de esta entidad con patologías pediátricas potencialmente fatales hace que su conocimiento sea de gran importancia para el pediatra general, puesto que el manejo temprano en la terapia intensiva es mucho más efectivo, disminuyendo la mortalidad por el SDRA.
Definición
La lesión pulmonar en el transcurso del tiempo ha variado desde la primera descripción de Ashbaugh en 1967, donde describe el síndrome de dificultad respiratoria del adulto en un grupo de 12 pacientes, uno de los cuales era un niño de 11 años de edad. El término dificultad respiratoria del adulto se tomó para diferencias esta entidad de el síndrome descrito en recién nacidos prematuros por deficiencia de surfactante. El diagnóstico de este síndrome requiere los siguientes elementos:
- Evento catastrófico pulmonar o extra pulmonar en paciente con pulmón previamente sano.
- Dificultad respiratoria con hipoxemia, disminución de la distensibilidad pulmonar e incremento de la fracción de cortocircuitos.
- Evidencia radiográfica de infiltrados pulmonares difusos.
- Exclusión de falla cardiaca izquierda o falla cardiaca congestiva.
En 1988 Murray y col, describen diferentes factores desencadenantes y datos clínicos en el síndrome de dificultad respiratoria del adulto, los cuales podrían dar inclusive el pronóstico de esta patología. Para el diagnóstico de esta patología se tienen que considerar las siguientes condiciones:
- Curso agudo o crónico
- Severidad de la lesión pulmonar. Cuadro # 1
- Causa o evento asociado (aspiración, sepsis, etc.).
El cambio de términos realizado de síndrome de dificultad respiratoria del adulto a síndrome de dificultad respiratoria aguda se realizó en el Consenso Americano Europeo de esta patología, en el cual se unificaron criterios para el diagnostico de esta entidad clínica, desechándose todas las definiciones anteriores y así poder manejar los mismos conceptos a nivel mundial. Sin embargo el término de SDRA es hasta cierto punto arbitrario, pues el SDRA denota la gravedad de la lesión pulmonar y en este mismo consenso se definieron los siguientes criterios:
- Inicio súbito de la sintomatología pulmonar
- Radiografía frontal de tórax con infiltrado bilateral
- Falta de evidencia clínica de hipertensión auricular izquierda o presión en cuña de la arteria pulmonar menor a 18 mmHg.
- El daño agudo pulmonar (DAP) requiere además relación PaO2/ FiO2 ≤ a 300 mm Hg
- Síndrome de dificultad respiratoria aguda relación PaO2/FiO2 ≤ 200 mm Hg.
En enero del 2000 Edward hace notar la necesidad de modificar estos criterios tomando en cuenta alteraciones fisiopatológicas y marcadores humorales de la lesión endotelial para poder diferenciar esta entidad de otro tipo de problemas que pueden tener los mismos datos clínicos, puesto que problemas inmunitarios o infecciosos pueden tener infiltrado pulmonar sin falla cardiaca y un inicio súbito.
Epidemiología
En 1972 el National Heart and Lung Institute estimaba una incidencia de 150,000 casos por año en los EUA, con una incidencia en la población total de 75/100,000 habitantes por año; estudios posteriores mostraron sin embargo una incidencia menor de aproximadamente 1,5 a 8,4 casos por 100,000 habitantes, posiblemente por el empleo de los nuevos criterios diagnósticos. La incidencia de esta patología en el niño, es aparentemente similar a la de los adultos, informandose en 1993 por Davis una incidencia de 12% en pacientes internados en terapia intensiva, con una mortalidad del 80% en los años 70 y que en los años 90 no mostró una disminución significativa. La alta mortalidad está determinada por la disfunción orgánica que se desencadena al tener una disminución en la disponibilidad tisular de oxígeno que da como efecto neto la lesión celular y la falla orgánica subsiguiente.
Etiología
Las causas que pueden ocasionar la lesión pulmonar que desencadena el DAP/SDRA son múltiples y Gatinoni las agrupa en causas de origen pulmonar y de origen extra pulmonar.
Las causas mas frecuentes de lesión de origen pulmonar son: la broncoaspiración, casi - ahogamiento, neumonía y neumonitis . Las causas de lesión de origen extrapulmonar son: la pancreatitis, sepsis, peritonitis, traumatismo craneano severo, politraumatismo y bomba de circulación extracorpórea, etc. La determinación del origen de la lesión pulmonar es de vital importancia tanto para el pronóstico como para el manejo ventilatorio.
Patología
Clásicamente se describen tres estadios en la evolución patológica del la lesión pulmonar que son la fase exudativa, fase proliferativa y fase de fibrosis. Estas etapas son mostradas en la gráfica uno.
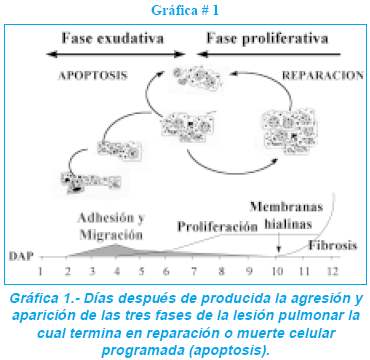
Fase exudativa. Esta fase se caracteriza por que su inicio esta comprendido dentro de las seis primeras horas de producida la lesión ya sea pulmonar o extra pulmonar, encontrándose congestión intraluminal, agregación de plaquetas, neutrófilos y fibrina en la luz de las arteriolas pulmonares que tienen de 25 a 250 micrómetros de diámetro. A las 12 a 24 horas de producido el insulto se encuentra marcada lesión periarterial y hemorragia intersticial con formación de membrana hialina en las siguientes 72 horas.
Las células endoteliales presentan edema y necrosis con destrucción de mitocondrias, sistema reticuloendoplásmico y ribosomas. Se encuentra además separación marcada de los endotelocitos por cambios en la estructura de la célula misma formando verdaderas brechas vasculares por donde se mantiene activo el proceso inflamatorio. Entre los mecanismos que explican la lesión endotelial tenemos:
- Agregación de neutrófilos y liberación de complemento.
- Lesión endotelial por liberación de productos de la coagulación en el espacio intravascular.
- Liberación de mediadores inflamatorios.
- Activación de otras células de inflamación como macrófagos y eosinofilos.
La lesión endotelial como se muestra en la gráfica # 1, en una primera instancia se observa la migración del neutrófilo hacia la pared del vaso sanguíneo, mucho mas manifiesto en el capilar pulmonar que es el más delgado de la economía humana, este neutrófilo al estar estimulado ya sea por lesión pulmonar o extrapulmonar libera mediadores inflamatorios y expresa moléculas de adhesión intercelular, estas moléculas al tener su contraparte en los endotelocitos estimulados producen en una primera instancia una adhesión intercelular débil por lo cual el neutrófilo empieza a “rodar” por la pared del capila,r si la lesión o el estimulo persiste se expresan moléculas de adhesión fuerte quedando la célula sanguínea anclada a la superficie del capilar pulmonar, en ese momento la liberación de mediadores inflamatorios es mucho mas intensa ampliándose la activación de neutrófilos.
Al producirse un intercambio de señales químicas intercelulares, se desencadena un acortamiento real del neutrófilo y liberación de las proteínas inter.-endotelocitarias creándose una brecha por la cual el neutrófilo puede emigrar al intersticio donde desencadena la respuesta inflamatoria que es responsable de la lesión pulmonar. Como se ve en la gráfica # 2, la respuesta desencadenada por el neutrófilo activa a las otras células de la inflamación que tienen como efecto la activación del endotelocito y la reacción vascular que inicia la secuencia fisiopatológica de la lesión pulmonar.
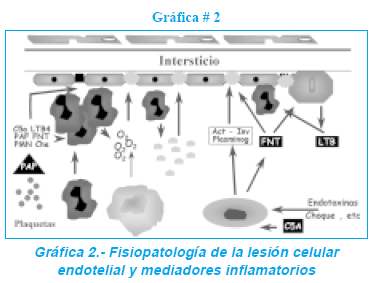
Todo este proceso desencadena la formación de exudados intra alveolares que son los responsables de la mala difusión de oxígeno a través de la membrana capilar que es la más perjudicada por esta activación celular, esta de mas decir que la activación de los neutrófilos y endotelocitos puede estar dada por procesos locales y distantes, pues el endotelio vascular funciona como un solo órgano en toda la economía del organismo humano.
Fase proliferativa. Esta fase ocurre entre la primera a tercera semana de producido el insulto, se caracteriza por la proliferación de neumocitos tipo II, fibroblastos y miofibroblastos. Los neumcitos tipo II recién formados tiene una estructura anormal, lo que denota que no son capaces de producir surfactante en suficiente cantidad, la función de estas células es la de formar nuemocitos tipo I que son los que cierran la lesión alveolar. Al mismo tiempo empiezan a proliferar fibroblastos tanto en la luz alveolar donde forman tejido de granulación el cual es sustituido por tejido fibrótico en la siguiente fase. La proliferación de fibroblastos además ocasiona la mala difusión de gases pues se produce una acumulación de fibrina en el intersticio que es parte de la membrana alvéolo capilar.
Fase de Fibrosis. Después de las tres semanas de producida la lesión el pulmón empieza a producir tejido colágeno para su remodelación y se inicia la angiogenesis que repara los vasos sanguíneos lesionados. La radiografía de tórax muestra fibrosis intersticial con formación de quístes pulmonares, lo que disminuye en gran medida la superficie de intercambio gaseoso.
Fisiopatología
Múltiples mecanismos son los responsables de la producción de la lesión pulmonar que ocasiona la mala difusión de gases a través de la membrana alvéolo capilar con la consecuente baja disponibilidad de oxígeno y muerte celular
Mecanismos de edema pulmonar. la difusión de líquido a través de una membrana semi permeable como es el endotelio, está regida por la ecuación de Starling que favorece la salida de algunos líquidos al intersticio y la entrada del agua al compartimiento intravascular la fórmula es la siguiente:
![]()
Donde Q es el índice de filtración a través de la membrana, K es el coeficiente de filtración, PC la presión hidrostática capilar, PIS la presión hidrostática insterticial, σ el coeficiente de reflexión, ![]() la presión oncótica plasmática y
la presión oncótica plasmática y ![]() presión oncótica intersticial.
presión oncótica intersticial.
La lesión pulmonar en el DAP/SDRA ocasiona la alteración de todos los componentes de ecuación pues el incremento de la presión de la arteria pulmonar por la congestión capilar determina una alteración de las presiones hidrostáticas y el coeficiente de difusión de los líquidos intravasculares se ve favorecido por las brechas celulares existentes, esto es mas evidente en el incremento de la concentración de proteínas en el intersticio pulmonar, donde condicionan la fuga de agua, el coeficiente de reflexión se ve alterado además por la perdida o variación de la carga eléctrica del endotelocito, favoreciendo la aproximación de proteínas polares a las cuales en situaciones normales no se podrían adosar a la superficie endovascular.
Mediadores de la inflamación. La respuesta inflamatoria montada en la lesión pulmonar en si es la misma que se produce en cualquier sitio de la economía donde las células proinflamatorias (polimorfonucleares, macrófagos y eosinófilos) son los responsables de esta alteración
Neutrófilos. Son las primeras células que llegan al sitio de la agresión y son los responsables de la producción de complemento C5a, leucotrieno B4, factor activador de plaquetas, factor de necrosis tumoral alfa e interleucina uno, todos estos factores estimulantes de la quimiotaxis de neutrófilos, por esta razón estos mismos son los que amplifican su propia respuesta además de estimular su adhesión al endotelocito. Además el factor de necrosis tumoral y la interleucina uno, parecen ser los responsables de la expresión de moléculas de adhesión de leucocitos por la célula endotelial.
Macrofagos. El desarrollo del SRDA en pacientes neutropénicos severos fue el primer dato para pensar que otras células podrían mediar la respuesta inflamatoria pulmonar encontrándose que el macrofago puede ser activado por C5a e interleucina uno, que a su vez es producida por esta célula, la cual produce además factor de necrosis tumoral alfa, la activación de estas citocinas puede desencadenar la lesión endotelial y en mayor grado la interleucina uno, la proliferación de fibroblastos que son los responsables de la tercera fase de la lesión pulmonar en esta patología.
Eosinófilos. Los eosinófilos por si solos pueden producir la liberación de radicales libres de oxígeno que son los mediadores de la destrucción celular en cualquier proceso inflamatorio y estos son encontrados en gran cantidad en el SDRA, hallándose también en lavado bronquial proteína cationica eosinofílica que demuestra su presencia y papel en la lesión pulmonar,.
Manifestaciones clínica y radiológicas
La lesión pulmonar en la clínica se manifiesta por la aparición de dificultad respiratoria en un individuo con pulmón previamente normal, es importante determinar la presencia de lesión pulmonar o lesión extrapulmonar seria que pueda justificar la aparición de esta patología, en general el deterioro ventilatorio se presenta dentro de las primeras seis horas de sufrido el insulto, con datos de dificultad respiratoria progresiva que se manifiesta por incremento del puntaje de Silverman y una presión arterial de oxígeno por debajo de 50, a pesar de una FiO2 mayor al 60’%, con una franca alteración del índice PaO2/FiO2.
La radiografía de tórax, demuestra presencia de infiltrado pulmonar difuso bilateral que no existía en radiografías previas, la imagen cardíaca es generalmente normal y el grado de infiltrado como se muestra en el cuadro # 1, es un dato que puede ser útil para catalogar el compromiso pulmonar. La radiografía de tórax puede ser determinante para la estadificación de la patología, encontrándose infiltrado difuso en la fase exudativa con formación de atelectasias en la proliferativa y fibrosis en la ultima fase de la lesión pulmonar.
Tratamiento
Dos son las terapias reconocidas para el manejo de esta patología ; la terapia ventilatoria y la farmacológica, la primera mucho más efectiva que la segunda que aún se encuentra en etapa de investigación.
Terapia ventilatoria. Aunque el SDRA ha sido considerado previamente un problema de daño pulmonar difuso y un incremento generalizado de tejido degenerado, actualmente parece que las consecuencias radiográficas, densitométricas y mecánicas son heterogéneas. En varios casos, la capacidad de distensión de los pulmones puede ser menor a un tercio del normal. La distensibilidad y fragilidad de los tejidos que comprenden los compartimentos aereados en el SDRA, parecen funcionar en forma normal, especialmente en la fase temprana de la enfermedad. La resistencia tisular y la vía aérea están elevados en el SDRA, una observación que tal vez es mejor explicada por el reducido número de unidades de la vía aérea. La hipoxemia refractaria del DAP puede elevarse supliendo el O2 inspirado y aumentando la presión alveolar al final de la espiración. Sin embargo, cada una de éstas intervenciones, se asocia a riesgos y beneficios. Estudios en animales han mostrado que altas fracciones de O2 inspirado y altas presiones cicladas son potencialmente dañinas, especialmente si se aplican durante períodos prolongados sobreponiéndose al daño preexistente, o combinándose con otros agentes.
Los objetivos de la ventilación, en el marco de DAP, han sido dar prioridad a normalizar los gases sanguíneos arteriales y evitar la depresión del gasto cardíaco. Hasta ahora, las presiones del sistema respiratorio en los humanos, han sido monitorizadas pero no explotadas al máximo. El flujo controlado, la ventilación volumen-ciclado utilizando volúmenes corrientes de 10 a 15 mL/kg, previamente han sido los estándares de la práctica en el manejo del SDRA.
La presión baja de vía aérea, y la retención de fluido bajo condiciones de inflación pasiva. La presión positiva al final de la espiración (PEEP) ha sido utilizada para incrementar la presión transalveolar y el volumen y por tanto mejorar el intercambio gaseoso. La presión alveolar que determina el volumen aereado al final de la espiración, es la suma de la PEEP aplicada deliberadamente y la cual puede ser aumentada por hiperinflación dinámica (auto-PEEP o PEEP intrínseco). Este último puede ser frecuentemente significativo en el DAP/SDRA, debido a su alta ventilación -minuto, al uso de tiempo inspiratorio largo, a la elevada resistencia de la vía aérea, tubo endotraqueal y problemas en la válvula de exhalación.
Todas las formas de barotrauma descritas en la literatura pediátrica, incluido el enfisema intersticial, quiste a tensión, embolismo aéreo sistémico y daño similar a la displasia broncopulmonar; han sido reconocidos en pacientes con SDRA.
En animales de experimentación la elección del patrón ventilatorio influye en la morfología del tejido normal previamente dañado. De los estudios de estos animales, se sospechó que volúmenes regionales excesivos son dañinos, ya sea por presión positiva o negativa. Por esta razón los volúmenes pulmonares tiene ser manejados muy juiciosamente.
Un estudio reciente sugiere que la sobredistensión regional esta comúnmente producida en pacientes con SDRA por una presión estática de la vía aérea mayor a 30 cm H2O; este nivel de presión causa daño cuando es mantenido por más de algunas horas. Aunque el volumen corriente excesivo debe ser evitado, estudios en animales sugieren que inflaciones periódicas con un volumen largo y sostenido pueden ser necesarias para evitar el colapso de unidades pulmonares inestables, cuando se utiliza volúmenes corriente pequeños (< 4-5 mL/kg). Parece ser que los pulmones son capaces de resistir a la exposición de algunas fuerzas elevadas en la fase temprana del SDRA en el humano, sin evidencia radiográfica de barotrauma. Más tarde, en el curso de la enfermedad, la fuerte infraestructura del colágeno de los pulmones, se degrada en forma dispareja.
Existe, además, incapacidad en preservar una presión transalveolar mínima al final de la espiración, en la fase temprana del SDRA; lo cual puede intensificar el daño alveolar preexistente, especialmente si se utiliza volúmenes corrientes elevados. La PEEP requerida para alejar un colapso alveolar esparcido varia con las fuerzas hidrostáticas aplicadas al pulmón. Consecuentemente, una alta presión es requerida en pacientes, para prevenir atelectasias, más en regiones pendientes que en regiones más superiores. Por tanto, los factores gravitacionales ayudan a explicar la distribución notablemente pendiente de los infiltrados radiográficos que siguen al daño pulmonar; así como la reversión de estos infiltrados y la mejoría de la oxigenación arterial en la posición prona.
El estrés de la incapacidad de los capilares pulmonares con el resultado de la extravasación de elementos formes de la sangre, pueden ocurrir a presiones transvasculares que excedan los 40 a 90 mmHg. Las fuerzas mecánicas transcapilares de magnitud comparable, pueden ser regeneradas en el SDRA cuando se utilizan volúmenes corrientes elevados y presión estática pico. Las presiones vasculares elevadas y el flujo sanguíneo también pueden ser importantes determinantes en el daño pulmonar.
Algunos coadyuvantes en la ventilación mecánica, como la inhalación del óxido nítrico, la insuflación del gas traqueal y la ventilación parcial líquida con perfluorocarbono asociado, mejoran el intercambio de gases transpulmonaes. La administración de surfactante y la prostaciclina inhalada, pueden mejorar el cuadro clínico.
Tomando en cuenta estos conceptos las recomendaciones dadas por el segundo consenso americano europeo de daño agudo pulmonar son:
- Asegurar la llegada apropiada de O2 a los órganos vitales mientras el CO2 se remueve en forma suficiente para mantener un equlibrio homeostático; aliviar el trabajo de respiración y evitar la extensión del daño pulmonar o prevenirlo.
- Minimizar la toxicidad por O2, utilizando una alta fracción de O2 inspirado a través de mascarilla por breves períodos como una medida temporal.
- A un cierto nivel de la ventilación minuto, la baja presión de la vía aérea puede incrementarse con la adición de la PEEP o extendiendo el tiempo de fracción inspiratoria. Aunque la reclutamiento alveolar puede continuar a través de volúmenes corrientes distintos, los valores de PEEP que obliteran la zona de infección más baja de la curva de inspiración estática presión-volumen, del sistema respiratorio, pueden asegurar un reclutamiento completo.
- Minimizar altas presiones de la vía aérea a través de la hipercapnea, ventilación con presión controlada y ventilación volumen-ciclado.
- Prevenir atelectasias utilizando grandes volúmenes, presión respiratoria elevada de inspiraciones de duración larga.
- Uso juicioso de sedación y relajación:
Terapia farmacológica. La comprensión de los procesos fisiopatológicos de la lesión pulmonar han llevado a buscar nuevas terapias para contrarrestar los efectos nocivos de la respuesta inflamatoria desencadenada a nivel pulmonar, inclusive la medición de estos mediadores inflamatorios y su cantidad en el torrente sanguíneo tratan de tomarse como factores predictores en la evolución y tratamiento de esta patología. En recientes trabajos se demostró sin embargo que el equilibrio entre los mediadores de la lesión celular, con los anti-inflamatorios como el glutation, son determinantes para montar una respuesta adecuada al desequilibrio entre la proinflamación y la respuesta contrainflamatoria puede derivar en la muerte del paciente.
Los medicamentos investigados son:
Corticoesteroides. En la fase inicial de la lesión pulmonar no es útil para disminuir la gravedad de la respuesta inflamatoria, ni tampoco es útil en pacientes sépticos en los cuales la lesión pulmonar se desarrolla a pesar de la corticoterapia, solamente se ha demostrado su utilidad en pacientes con neumonía por pneumocistis carini en los cuales la administración de corticoides diminuye la incidencia de SDRA, también es útil su uso en la tercera fase de la lesión pulmonar donde la proliferación fibroblástica es la responsable de la lesión pulmonar cicatrizal.
Pentoxifilina. Este fármaco es un derivado de las xantinas que tiene varias acciones a nivel de la circulación pulmonar pues disminuye la resistencia de la arteria pulmonar, inhibe los radicales libres de oxígeno, el factor activador de plaquetas y por ende la agregación plaquetaria y la fagocitosis además de su conocido efecto homeorético en los glóbulos rojos. Este fármaco puede ser útil para el manejo de esta patología, actualmente se encuentra en estudio una nueva molécula que es la lisofilina que podría ser de mayor utilidad.
Remplazo de surfactante. Al presentarse membranas hialinas al inicio de la lesión pulmonar la terapia con surfactante siempre fue una opción en el manejo del SDRA, sin embargo la gran cantidad de fármaco que se requiere para niños mayores o adolescentes elevan su costo además que su poca utilidad es el principal limitante para utilizarlo; aunque existen resultados alentadores con la terapia temprana.
Terapia génica. Se encuentra en etapa de experimentación la aplicación de genes para la producción de alfa 1 antitripsina, MnSOD y prostaglandina G/H sintetasa, y se está probando como activar y desactivar estos genes y su administración pulmonar.
Otros tratamientos se detallan en la tabla # 1, siendo la mayoría de estos aun experimentales y con resultados en algunas ocasiones contradictorios.
Conclusiones
La lesión pulmonar ya sea daño agudo o síndrome de dificultad respiratoria aguda, es aún un desafió en cuanto a su tratamiento se refiere, sin embargo la prevención y anticipación a la presentación de la lesión pulmonar es de suma importancia tanto para el tratamiento como para el pronóstico. Además que la uniformidad de criterios para el diagnóstico de esta patología facilitan su manejo y así poder aplicar protocolos de investigación e introducir terapias potencialmente beneficiosas.
Referencias
1. Bernard GR, Artigas A, Brigham KL, Carlet K, Flake L, Hudson L, et al. The American-European Consensus Conference on ARDS: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Intensive Care Med 1994; 20:225-32. [ Links ]
2. Artigas A, Bernard GR, Carlet J, Dreyfuss D, Gattinoni L, Hudson L. et al: The American-European Consensus Conference on ARDS, Part 2. Ventilatory, pharmacologic, supportive therapy, study design strategies and issues related to recovery and remodeling. Intensive Care Med 1998; 24:378-98 [ Links ]
3. Brower RG, Ware LB, Berthiaume Y, Matthay MA. Treatment o ARDS. Chest 2001 on line 120 (4). [ Links ]
4. Clark RH, Gerstmann DR, Jobe AH, Moffitt ST, Slutsky AS, Yoder BA. Lung injury in neoates: causes, strategies for prevention, and long-term consequences. J Pediatr 2001:139(4) on line [ Links ]
5 Fein AM, Calalang MG. Acute lung injury and acute respiratory distress syndrome in sepsis and septic shock Critical Care Clin 2000;16(2) on line [ Links ]
6. Hudson LD Steinberg P. Epidemiology of acute lung injury and ARDS. Chest 1999: 116: 75 – 82.
7. Ingbar DH. Mechanisms of repair and remodeling following acute lung injury. Clin Chest Med 2000; 21(3): on line [ Links ]
8. Krishnan JA, Brower RG. High-frequency ventilation for acute lung injury and ARDS. Chest 2000; 118(3): on line [ Links ]
9. Matthay MA, Dinarello CA, Vincent JL, Cohen J, Opal SM, Glauser M, Parsons P, Fisher CJ, Repine JE. Consensus Conference definitions for sepsis, septic shock, acute lung injury, and acute respiratory distress syndrome: time for a reevaluation Critical Care Med 2000; 28:232-5. [ Links ]
10. McIntyre RC, Pulido EJ, Bensard DD, Shames BD, Abraham E. Thirty years of clinical trials in acute respiratory distress syndrome. Critical Care Med 2000; 28: 3314–30.
11.Mitchell RS, Martin TR. Lung cytokines and ARDS. Chest 1999; 116:1–8.
12. Parsons PE. Mediators and mechanisms of acute lung injury. Clin Chest Med 2000; 21(3): on line [ Links ]
13. Smith PG, Blumer J. NO good--or not? Critical Care Med 1999: 27:1059–60.
14. Tomashefski JF. Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med 2000; 21 (3): on line [ Links ]
15.Wiedemann HP. Partial liquid ventilation for acute respiratory distress syndrome. Clin Chest Med 2001: 21 (3): on line [ Links ]

