










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Boliviana de Pediatría
versión On-line ISSN 1024-0675
Rev. bol. ped. v.41 n.1 La Paz ene. 2002
CASO CLINICO
Hipotiroidismo congénito. A propósito de un casoCongenital hypothyroidism
Dres.: Manuel Pantoja Ludueña*, Ac. Eduardo Mazzi Gonzales de Prada*, Kurt Paulsen Sandi**
* Médico Pediatra, Servicio de Neonatología, Hospital del Niño “Dr. Ovidio Aliaga Uría”.
** Médico Pediatra, Servicio de Urgencias, Hospital del Niño “Dr. Ovidio Aliaga Uría”.
Dirección: Calle My Zubieta s/n. Miraflores.
La Paz Teléfono: 2240081. Email: mpantoja@ceibo.entelnet.bo
Resumen
El hipotiroidismo congénito es la enfermedad endocrinológica mas frecuente en pediatría y a su vez es la primera causa de retardo mental prevenible. El diagnóstico precoz se establece mediante el cribado neonatal de los niveles de hormona tiro-estimulante TSH, puesto que solamente un pequeño porcentaje de niños presenta sintomatología clínica en el periodo neonatal.
Se describe el caso clínico de un recién nacido atendido en la sala de neonatología del Hospital del Niño “Dr. Ovidio Aliaga Uría”, a quien se le diagnosticó hipotiroidismo congénito a los 11 días de vida y se empezó tratamiento de sustitución con levo tiroxina sódica, para evitar las secuelas neurológicas posteriores. El seguimiento de paciente durante 5 meses demuestra un desarrollo normal.
Se presenta el caso para resaltar la importancia del diagnóstico precoz y la utilidad de implementar el cribado neonatal en forma universal.
Palabras Claves:Rev. Soc. Bol. Ped. 2002; 41 (1): 11-14: Hipotiroidismo congénito, retardo mental, cribado neonatal, TSH neonatal.
Abstract
Congenital hypothyroidism is one of the most frequent endocrinological disease in pediatrics and the most common cause of mental retardation. It is important the diagnosis thru the routine screening of neonatal TSH, because most of the newborn are asymptomatic.
We describe an 11 day-old infant seen at the Hospital del Niño “Dr. Ovidio Aliaga Uría”, the follow up for 5 months shows a good response to treatment.
We emphasize the importance of the routine screening for the disease and the importance of the early treatment.
Key words:Rev. Soc. Bol. Ped. 2002; 41 (1): 11-14: Congenital hypothyroidism, mental retardation, neonatal screening, neonatal TSH.
Introducción
El hipotiroidismo congénito (HC) se define como una insuficiencia tiroidea presente desde el nacimiento debido a la ausencia de la glándula tiroidea o falta de acción de hormonas tiroidea durante la vida fetal. Si se desarrolla hipotiroidismo desde la etapa fetal se afecta principalmente el desarrollo del sistema nervioso central y esquelético, aún así, la mayoría de los recién nacidos afectados parecen normales debido a la protección relativa y transitoria otorgada por el paso transplacentario de hormonas tiroideas maternas. Es la enfermedad endocrina congénita mas frecuente en pediatría y a su vez es la causa mas común de retardo mental prevenible por medio de un diagnóstico temprano y tratamiento oportuno.
La prevalencia del HC es aproximadamente 1 caso por cada 4000 recién nacidos vivos, sin embargo esta prevalencia se ve influida por factores genéticos. Así, la incidencia es mas elevada en las etnias orientales e hispanas con 1 caso por cada 2000; es menor en la etnia blanca con 1 caso por cada 5500 y bastante más baja en la etnia negra, con 1 caso por cada 32000 estudios recientes demuestran un porcentaje elevado de anomalías tiroideas en los parientes de pacientes con HC, que indica una transmisión autosómica dominante de baja penetración. En nuestro país no tenemos estadísticas propias puesto que no se ha implementado un programa de nacional de detección de esta enfermedad.
La única manera de hacer un diagnóstico precoz de HC es a través del cribado neonatal, dosificando la hormona tiro-estimulante a partir del segundo día de vida. Programa en funcionamiento desde 1972 en los países desarrollados y gran partes de los países en desarrollo.
A continuación presentamos el caso clínico de un recién nacido atendido en la sala de neonatología del Hospital del Niño “Dr. Ovidio Aliaga Uría”, a quien se le diagnosticó HC y aprovechamos la oportunidad para hacer una revisión del tema.
Caso clínico
Recién nacido femenino de 11 días de vida, producto del tercer embarazo, parto domiciliario, madre de 27 años de edad quien desarrollo tiroiditis de Hashimoto al mismo tiempo que se detectó el HC a hijo; alimentado con lactancia materna exclusiva. Fue traído al hospital del Niño “Dr. Ovidio Aliaga Uría”de La Paz, por presentar ictericia de ocho días de evolución, somnolencia, succión débil, distensión abdominal y constipación. Al examen físico se destacó un peso de 4100 gramos, talla de 52 centímetros y perímetro cefálico de 37.5 centímetros. Presencia de ictericia que comprometía las palmas de las manos y plantas de los pies, fontanela posterior grande de aproximadamente 1,5 centímetros de diámetro, abdomen distendido con 39 centímetros de circunferencia y presencia de una hernia umbilical pequeña.
Ante la sospecha de un hipotiroidismo congénito, se solicitó determinación de TSH , cuyo resultado fue de 483 uUI/ mL, con este resultado se confirmó la sospecha diagnóstica y se inició terapia de sustitución con levo-tiroxina sódica a una dosis de 8 ug/K/día, previa determinación sérica de hormonas tiroideas (TSH, T3 y T4) y centellografía tiroidea con tecnecio 99. Los niveles hormonales tiroideos se observan en el cuadro 1.

La centellografía mostró ausencia de concentración del indicador radioactivo en el lecho tiroideo y presencia de una pequeña zona de concentración a nivel sublingual con tejido tiroideo muy hipoplásico, figura 1.
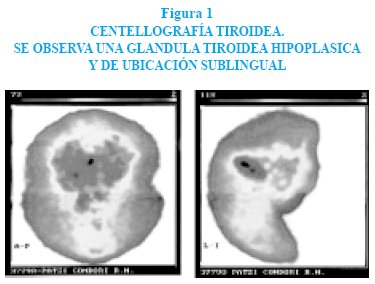
La hiperbilirrubinemia a predominio indirecto con valores totales de 22,3 m/dL y con bilirrubina indirecta de 20,7 mg/ dl, respondió a la fototerapia al segundo día de tratamiento.
En su primer control postratamiento, se observó el nivel de TSH levemente elevado por lo que se subió la dosis de levotiroxina a 11,7 ug/K/día, a los 20 días de este cambio se controlan nuevamente las hormonas tiroideas y niveles posteriores se observan el cuadro 1.
La evolución es favorable, la TSH se normalizó a los 45 días de tratamiento, la evaluación física y neurológica a los 2 meses de edad fue normal y el paciente acude a sus controles periódicamente.
Discusión
El hipotiroidismo congénito puede ser permanente o transitorio, como se observa en el cuadro 2. El 80 a 90% de los casos de hipotiroidismos permanentes son debidos a disgenesias tiroideas, ya sea por ausencia de la glándula (atireosis) o ectopia tiroidea con tejido hipoplásico, como el caso que describimos. Recientemente se ha demostrado, que la mutación del gen receptor de la TSH podría condicionar HC con hipoplásia de la glándula en grados variables. La transferencia de anticuerpos tiroideos de la madre al niño y una deficiencia moderada de yodo son las causas de hipotiroidismo congénito transitorio mas frecuentes. Este grupo de pacientes tienen un a incidencia variable, dependen del aporte de yodo a la población y en algunos casos el HC puede ser tan profundo que necesitan tratamientos prolongados con levo – tiroxina.

La ausencia de hormonas tiroideas producen en el sistema nervioso central un retardo en la arborización dendrítica, vascularización, migración neuronal y maduración de las conexiones interneuronales, que se traduce en lesiones irreversibles del tejido neuronal, produciendo retardo mental y otras alteraciones neurológicas. A nivel sistémico interfiere con todos los procesos metabólicos y de maduración del organismo sobre todo en el tejido óseo y por tanto en el crecimiento linear.
El crecimiento y desarrollo fetal es independiente al estado tiroideo. La participación de las hormona tiroideas en el crecimiento son importantes en el periodo postnatal donde se ha demostrado un sinergismo en la síntesis de hormona de crecimiento y tiroidea; por lo que se deduce que el feto con HC nace con talla normal que luego se va comprometiendo.
Las glándulas tiroidea y pituitaria fetal se producen entre la 10º y 12º semanas de gestación, pero son relativamente inactivas y comienzan un incremento gradual hacia el término del embarazo. Inmediatamente después del parto, el nivel de T3 se incrementa por la conversión de T4 a T3 en los tejidos y también se eleva la TSH neonatal con un máximo a los 30 minutos para luego descender gradualmente en las primeras 24 horas. El eje hipotalámico-hipofisiario-tiroideo fetal funciona en forma independiente del sistema materno y la placenta es impermeable a la TSH materna y relativamente impermeable a las hormonas T3 y T4 maternas, aunque existe algún paso transplacentario de T4 al feto que parece ser importante para mantenerlo eutiroideo en forma transitoria, es por eso que en el feto atiroideo o con ectopia tiroidea, a pesar de tener una TSH elevada, el crecimiento somático y el desarrollo neurológico transcurren normalmente y cualquier efecto de la deficiencia de hormona tiroidea fetal sobre la maduración cerebral parece ser reversible si la sustitución tiroidea posnatal se establece precozmente. Todo esto se cumple siempre y cuando la función tiroidea de la mujer embarazada sea normal, en el caso de una madre hipotiroidea gestante se le debe aumentar la dosis de sustitución tiroidea entre el 30 a 50%.
La deficiencia de hormona tiroidea en los primeros años de vida produce cambios anatomofuncionales permanentes como ser: reducción del tamaño absoluto del cerebro y cerebelo, disminución de la capacidad de migración y proliferación de las células gliales, retrazo en la mielinización, deficiencia en la conducción axonal, disminución de las arborizaciones dendríticas, atrofia de las circunvoluciones y retraso psiconeurológico de diferente magnitud.
La mayoría de los recién nacidos con hipotiroidismo congénito no presentan sintomatología clínica típica de deficiencia alguna y solo en el 3 a 5% se puede sospechar clínicamente el diagnóstico, por lo que se dificulta la detección temprana de la enfermedad. En el caso de nuestro paciente, lo que hizo sospechar el diagnóstico fue la presencia de una fontanela posterior amplia, hernia umbilical e ictericia prolongada en un recién nacido del sexo femenino. Describimos los signos y síntomas del HC en el cuadro 3.
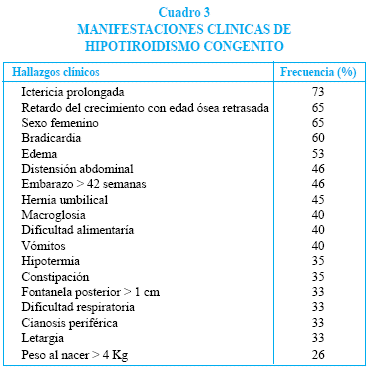
El diagnóstico se lo realiza por medio de la determinación sérica de las hormonas tiroideas donde se observa elevación de TSH y disminución de T4, aunque durante las primeras semanas o meses de vida el T4 puede estar normal en caso de tiroides ectópico o hipoplásico. En nuestro caso confirmamos los niveles elevadísimos de TSH y bajos de las hormonas tiroideas.
Antes de la era del cribado neonatal con TSH, se conocía que los factores pronósticos más importante eran el diagnóstico y tratamiento precoces. Estudios realizados antes de 1975 observaron que más del 50% de casos eran diagnosticados después de los tres meses de edad y la mayoría de los niños presentaban secuelas neurológicas serias. Este problema se resolvió con la introducción del cribado sistemático de TSH neonatal.
Para la pesquisa rutinaria del TSH neonatal se obtiene una muestra de sangre por punción del talón al segundo o tercer día de vida en los recién nacidos a término y al quinto día de vida en los prematuros, impregnándose un papel filtro especial por ambos lados, colocándo varias gotas separadas en un espacio de aproximadamente un centímetro de diámetro cada una. La dosificación de TSH neonatal se realiza por métodos de inmunoensayo, radioinmunoanalisis (RIA) o radioinmunometría (IRMA).
Todo recién nacido con valores de TSH neonatal superiores a 20 UI/mL debe ser considerado sospechoso de HC aún cuando no existan manifestaciones clínicas y debe confirmarse el diagnóstico mediante la evaluación completa de la función tiroidea, determinando niveles séricos de TSH, T3 y T4 y se recomienda iniciar tratamiento de sustitución hormonal hasta documentarse o descartarse la enfermedad.
Se recomienda determinar la edad ósea para evaluar la eficacia del tratamiento y obtener una centellografía tiroidea con tecnecio 99 para determinar la presencia de glándula tiroidea, que en el caso que presentamos descubrió una tiroides ectópica. Para evaluar la edad ósea se toman radiografías de la rodilla hasta los 3 meses de edad y posteriormente de la mano izquierda.
La centellografía tiroidea con tecnecio 99 es útil para ubicar la glándula tiroidea, como el caso de nuestro paciente. Otros exámenes complementarios que se pueden realizar son: ecografía tiroidea, captación tiroidea con yodo 131, medición de tiroglobulina, proteínas transportadoras, inmunoglobulina antitiroidea y determinación de anticuerpos para receptores de TSH, según cada caso en particular.
El objetivo del tratamiento es evitar el daño neurológico que es irreversible, mediante el tratamiento precoz, idealmente antes de los 15 días de vida. La droga de elección es la levo-tiroxina sódica, que por su vida media prolongada, se logra una mejor regulación de la TSH. La dosis inicial recomendada es de 10 µg/kg/día (7-15 µg/kg/día) en una sola toma oral diaria, con el objetivo de normalizar la TSH y T4 en 2 a 3 semanas. Posteriormente se modifica la dosis de acuerdo a la respuesta y niveles plasmáticos de T4 y TSH. Se recomienda mantener el nivel sérico de T4 debe por encima del rango normal de 10 a 16 g/dL, la T3 en rangos normales y la TSH en 5 UI/mL o ligeramente elevado.
Mientras más temprano se inicie el tratamiento, mejor será el coeficiente intelectual obtenido. Por ello, cuando estamos frente a un paciente con valores hormonales confusos, es preferible iniciar tratamiento hasta que se realicen otras pruebas para confirmar la enfermedad.
El tratamiento del HC verdadero es permanente, por lo tanto los padres deben estar conscientes de la enfermedad de su hijo y ser constantes en la administración de la terapia; en el caso de un HC transitorio debe mantenerse hasta los 2 años de edad, momento en que se evalúa la necesidad de proseguirlo a través de un estudio completo de la función tiroidea.
Los factores que ensombrecen el pronóstico son valores muy elevados de TSH neonatal, como el caso de nuestro paciente y retraso de la edad ósea. Estos dos factores denotan compromiso fetal importante e influyen negativamente en el desarrollo del coeficiente intelectual, maduración esquelética y función motora.
Por todo lo expuesto, recalcamos la importancia de introducir el cribado neonatal en forma sistemática en todas las maternidades del país.
Referencias
1. Abodovsky N. Hipotiroidismo congénito. En: Meneghello J, Fanta E, Paris E y Puga T, eds. Pediatría. 5ta ed. Buenos Aires: Editorial Panamericana;1997.p.1878-82. [ Links ]
2. American Academy of Pediatrics. Newborn screening for congenital hypothyroidism: recommended guidelines. Pediatrics 1993:91;1203-9. [ Links ]
3. Aznarez A, Balter H, Bardcia S, Díaz J, Iturralde A, Mañe F y col. Detección sistemática del hipotiroidismo congénito en todos los recién nacidos. Arch Pediatr Uruguay 1994; 65:47-51. [ Links ]
4. Barrón-Uribe C y Pérez-Pastén E. Conceptos actuales en hipotiroidismo congénito. Bol Med Infant Mex 1996; 53:264-8. [ Links ]
5. Burrow GN, Fisher DA, Larson PR. Maternal and fetal thyroid function. N Eng J Med 1994; 331:1072-8. [ Links ]
6. Di George A y La Franchi S. Hipotiroidismo. En: Behrman R, Kliegman R y Arvin A. Eds. Nelson. Tratado de pediatría. 15º ed.Madrid: McCraw-Hill Interamericana; 1997.p.2085-91. [ Links ]
7. Fisher D. Hypothyroidism. Pediatr Rev 1994; 15:277-32. [ Links ]
8. García J. Función tiroidea materna y la salud del niño. Arch Pediatr Urug 2001; 72:91-92. [ Links ]
9. Léger J, Marinovic D, Garel C, Boaníti C, Polak M, Czernichow P. Thyroid developmental anomalies in first degree relatives of children with congenital hypothyroidism. J Clin Endocrinol Metabolism 2002; 87:575-80. [ Links ]
10. Pantoja M, Mazzi E, De Avila R, Diaz M, Barragan D, Córdoba J. Hipotiroidismo congénito: propuesta de una norma para su detección temprana. Rev Soc Bol Ped 1996; 35:32-5. [ Links ]
11. Polk D. Diagnosis and management of altered fetal thyroid status. Clin Perinatol 1994; 21:647-61. [ Links ]
12. Scott M. Thyroid disorders. In: Cloherty J and Stark A, eds. Manual of neonatal care. 3º ed. Boston: Little Brown and Co.;1998.p.20-6. [ Links ]
13. Simmons WF, Fuggle PW, Grant DB, Smith I. Intellectual development at 10 years in early treated congenital hypothyroidism. Arch Dis Child 1994; 71:232-4. [ Links ]
14. Toft A. Thyroxine therapy. N Engl J Med 1994; 331:174-8. [ Links ]
15. Willi S, Moshang T. Resultados de las pruebas de detección para hipotiroidismo congénito. Clin Pediatr Norte América 1991; 38:565-77 [ Links ]

