











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
El SARS-CoV-2 contiene cuatro tipos de proteínas estructurales, entre ellas, la proteína de la espiga (S), las proteínas de la envoltura (E), las proteínas de membrana (M) y las proteínas de la nucleocápside (N)1,2. La infección viral por SARS-CoV-2 depende de la proteína S, que es una glucoproteína compuesta por subunidades S1 y S2, la cual se une a los receptores celulares ACE2 (receptor de la enzima convertidora de angiotensina 2) del huésped mediante el Dominio de Unión al Receptor (RBD) de la subunidad S1, para posteriormente, ingresar a la célula a través de su fusión con la membrana celular, proceso favorecido por la subunidad S23.
RBD es un blanco crítico para la unión tanto de compuestos antivirales, como de anticuerpos neutralizantes (Letko et al., 2020). SARS-CoV-2 RBD está formado por cinco hojas-β anti paralelas conectadas por bucles y hélices cortas. Entre estas cadenas anti paralelas se encuentra el Motivo de Unión al Receptor (RBM) cuya superficie es ligeramente cóncava (hacia adentro) para dejar espacio para el receptor ACE2, además, contiene la mayoría de los sitios de unión para este receptor4.
En Bolivia, desde que los primeros casos de SARS-CoV-2 fueron detectados a inicios de 2020, se produjeron hasta el momento, seis distintas olas de contagio, siendo las dos primeras olas producidas por la variante original de SARSCoV-2, mientras que las últimas cuatro, fueron a causa de variantes epidemiológicas que mutaron para transformarse en agentes mucho más contagiosos y evasivos para el sistema inmune, entre ellas, las variantes delta, omicron, centaurus y kraken, respectivamente5.
Desde que la OMS autorizó la administración de vacunas (noviembre, 2021), en Bolivia, las vacunas de Sinopharm y Sputnik V fueron algunas de las más aplicadas con 3 300 000 y 1 235 000 dosis, respectivamente6,7. La vacunación genera IgG anti-RBD que evita la entrada del virus a la célula, estos anticuerpos neutralizantes (AcN) son considerados un buen marcador de respuesta humoral y su actividad puede medirse por varios métodos8. Sin embargo, debido a las mutaciones producidas en el genoma de SARS-CoV-2 a lo largo de la pandemia, que dio lugar a la generación de diferentes variantes y sub-variantes, la eficacia de las vacunas ha sido constantemente evaluada, incrementando el número de dosis (hasta 5 en Bolivia), mientras que la población actualmente padece de reinfecciones constantes que aún ponen en riesgo a los grupos etarios más vulnerables9.
Actualmente, las herramientas bioinformáticas y los estudios clínicos han jugado un rol trascendental en el seguimiento de las todas las modificaciones y sustituciones en la secuencia de la proteína S (spike) de SARS-CoV-2, que la convirtieron en distintas variantes de importancia clínica con capacidades de transmisión y evasión inmunológica mucho más altas que la original reportada en Wuhan, China. Es muy probable, que por medio de este tipo de aplicaciones moleculares in-silico, podamos monitorizar y predecir anticipadamente los potenciales riesgos de nuevas variantes emergentes de SARS-CoV-2 en nuestro país.
Material y métodos
Alineamiento Múltiple de Secuencias (MSA)
Las secuencias del Dominio de Unión al Receptor (RBD) de la espiga de SARS-CoV-2 wild type y de todas las variantes analizadas en este trabajo, fueron obtenidas de Protein Data Bank (PDB: https://www.rcsb.org/): 6lzg_wt, 7ekf_alpha, 7ekg_beta, 7ekc_gamma, 7w98_delta, 7t9l_omicron, 7ykv_ centaurus y 8iov_kraken. Las secuencias correspondientes a RBD fueron determinadas con Chimera1. El MSA fue realizado con el software Mega6, ajustando el umbral esperado a 0,05, con una penalización para gaps de 11 y empleando BLOSUM80 como matriz de sustitución para aminoácidos.
Comparación Estructural de variantes RBD-SARS-Cov-2
Como estructura de referencia se empleó la estructura tridimensional correspondiente al Dominio de Unión al Receptor (RBD) de la espiga de SARS-CoV-2 wild type (región que va desde 333 a 527 aa), cuya distribución espacial fue comparada con todas las variantes analizadas en este estudio (alpha, beta, gamma, delta, omicron, centaurus y kraken). Se obtuvo el par de cadenas mejor alineadas incluyendo el alineamiento de las estructuras secundarias hasta que ninguno de los pares de cadenas exceda los 2.0 Ǻ de distancia entre sí. Para determinar el grado de similitud en la distribución espacial de los átomos en las estructuras tridimensionales, se calculó la raíz cuadrada media (RMSD) de las posiciones atómicas, que es la distancia promedio entre los átomos de las cadenas principales de las proteínas superpuestas estructuralmente. Valores cercanos a 0 indican que las estructuras comparadas tienen un alto grado de similitud en su distribución espacial.
Interacción de variantes RBD-SARS-Cov-2 con IgG neutralizante
Para el análisis comparativo se utilizó la estructura cristalográfica del dominio de unión al receptor (RBD) de SARSCoV-2 en complejo con el anticuerpo neutralizante CC12.1 (de tipo IgG específico para RBD) (PDB: 6xc2). A partir de este complejo, se realizaron las superposiciones estructurales del anticuerpo neutralizante CC12.1 con todas las variantes de SARS-Cov-2 analizadas en este trabajo, basándonos en los mismos criterios de alineamiento estructural mencionados en la anterior sección. Para encontrar posibles impedimentos estéricos y/o contactos entre átomos, se identificaron todos los átomos que estuvieran cercanos entre sí con un radio menor o igual a 0,6 Ǻ, que es una distancia muy por debajo del radio de Van der Waals permitido para el átomo de Hidrógeno (el más pequeño), de esta forma poder encontrar los conflictos estéricos de mayor importancia.
Parámetros cuantitativos
Para el Alineamiento Múltiple de secuencias de todas las variantes y sub-variantes de SARS-CoV-2 desarrolladas en este trabajo, se calculó el % de identidad, el MaxScore y el E-value, como parámetros cuantitativos. Para la comparación estructural del RBD de SARS-CoV-2, se calculó el RMSD (Desviación de la raíz cuadrada media) de las posiciones atómicas. El análisis de la complementariedad de las variantes y sub-variantes de RBD de SARS-CoV-2 con el AcN IgGantiRBD se empleó la función del alineamiento estructural en Chimera 1.12 para calcular el número de todos los posibles impedimentos estéricos entre los átomos que participan en la interacción Ag-Ac.
Resultados
RBD_Kraken tiene el mayor % de variación
En el Alineamiento Múltiple de Secuencias de aminoácidos se observa que el Dominio de Unión al Receptor (RBD) de la espiga de SARS-Cov-2 perteneciente a la variante omicron y sus sub-variantes centaurus y kraken presentan un mayor número de aminoácidos mutados en comparación con la secuencia wild type (RBDwt SARS-CoV-2) (Figura 1). Estos aminoácidos mutados, se encuentran distribuidos a lo largo de toda la secuencia del RBD (333 - 527aa), sin embargo, las principales mutaciones se encuentran localizadas en las regiones que posiblemente afectan la eficacia en la unión con los anticuerpos neutralizantes IgG (Figura 1, recuadro rojo).

Figura 1. MSA de la secuencia del RBD (333-527aa) de SARS-CoV-2 wt y las diferentes variantes de importancia clínica. Las mutaciones sinónimas y no sinónimas están señaladas con flechas verdes y rojas, respectivamente. Recuadro rojo: secuencia de interacción con IgG-antiRBD. Recuadro anaranjado: Sustituciones que producen impedimentos estéricos. Recuadro verde: sustituciones que mejoran la unión con ACE2 (Elaboración propia a partir de la base de datos del NCBI y el software Mega6).
Los parámetros cuantitativos como el E-value y %Identity basados en la matriz de sustitución BLOSUM80 (Tabla 1), señalan que la variante kraken es la que más difiere de su secuencia original.
Tabla 1. Parámetros cuantitativos del MSA (basados en BLOSUM80) entre RBD_SARS-CoV-2 wt y las diferentes variantes.

Fuente: bases de datos de la investigación
RBD_Omicron es la más divergente a nivel estructural
La comparación estructural nos muestra que de acuerdo al valor calculado de RMSD, la variante con el valor RMSD más alto (1,934) corresponde a la estructura del RBD de omicron, siendo la que presenta más diferencias a nivel estructural en comparación con RBD de SARS-CoV-2wt (Figura 2).
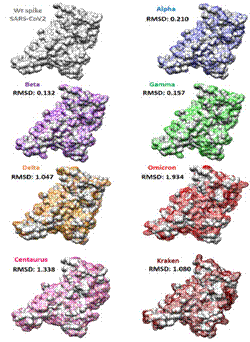
Figura 2. Alineamiento Estructural entre RBD de SARSCoV2wt (la primera cristalizada en Wuhan) y las diferentes variantes (Elaboración propia a partir de la base de datos PDB y el software Chimera1.15).
RBD_Omicron es la de menor complementariedad con AcN IgG-antiRBD
IgG-antiRBD interacciona con RBD de SARS-CoV-2wt formando 8 puentes de H2, sin embargo, el alineamiento estructural señala una disminución en los puentes de H2 cuando este anticuerpo neutralizante interacciona con la variante omicron (Figura 3). Por otra parte, el análisis de la superficie molecular de interacción nos muestra que la interacción entre IgG con la variante omicron produce 167 contactos no permitidos con un radio menor a 0,6 Ǻ evidenciando la presencia de varios choques entre sus átomos (impedimentos estéricos) (Figura 4)

Figura 3. Puentes de H2 (líneas amarillas) en la interacción de IgG-antiRBD con RBD de SARS-CoV-2wt y omicron (Modelo de cintas) (Elaboración propia, PDB: 6xc2, software Chimera).

Discusión
Desde que fue declarada la pandemia, hasta la actualidad, en la que se han desarrollado y aplicado diferentes tipos de vacunas en todo el mundo, el virus denominado como SARSCoV-2 ha sufrido un proceso de mutación en su genoma, que le ha permitido dar origen a distintas variantes y sub-variantes que han ocasionado nuevos brotes de contagio. Entre las principales variantes registradas durante todo este periodo (2019 - 2023) se encuentran las variantes alpha, beta, gamma, delta, omicron y sus sub-variantes centaurus y kraken5.
Muchas de las mutaciones introducidas en el genoma de SARS-CoV-2, produjeron un cambio en la secuencia de aminoácidos de diferentes proteínas virales, sin embargo, las de mayor importancia clínica fueron los cambios producidos en la proteína S (spike) que es la encargada de interaccionar con los receptores ACE2 para el ingreso del virus a las células, y también es el sitio de reconocimiento antigénico por el sistema inmune, más específicamente, la región de la proteína S denominada como Dominio de Unión al Receptor (RBD) el cual también es el sitio de unión de los anticuerpos neutralizantes (AcN) IgG-antiRBD, que proporcionan inmunidad contra este virus4,10,11.
El resultado del Alineamiento Múltiple de Secuencias señala que las principales mutaciones no conservadoras se encuentran en la región del Dominio de Unión al Receptor (RBD), siendo la sub-variante de omicron conocida como kraken, la que presentó un mayor número de mutaciones no conservadoras en su secuencia de aminoácidos, sin embargo, el grado de divergencia estructural calculado con base al valor RMSD nos indica que omicron es la variante con mayor divergencia estructural, debido a que no toda mutación en la secuencia de aminoácidos necesariamente se traduce en una alteración en la estructura tridimensional de la proteína. Como consecuencia, la variante omicron tuvo una reducción marcada en su capacidad de formar puentes de H2 y un incremento notable en el número de posibles impedimentos estéricos al interaccionar con anticuerpos neutralizantes (IgGantiRBD). Si realizamos una comparación de la estructura de cada aminoácido sustituido (Figura 1), podemos evidenciar que las sustituciones que favorecen impedimentos estéricos son los que sustituyen un aminoácido con cadena lateral pequeña a otro con una cadena lateral más voluminosa, como es en el caso de las sustituciones N501Y, Q498R, N460K y G496S. Aunque no se tomaron en cuenta otros tipos de interacciones como las iónicas, hidrofóbicas o de tipo Van der Waals; el valor elevado de RMSD, la reducción marcada en los puentes de hidrógeno y el alto número de átomos con contactos no permitidos entre RBD y AcN IgG, nos indican que la variante omicron podría ser la variante más evasiva para el sistema inmune en la población vacunada. Aunque en este trabajo no se realizó un análisis de la interacción de SARS-CoV-2 con los receptores ACE2, estudios recientes indican que la mutación N439K (presente en omicron, centaurus y kraken) incrementa su afinidad por el receptor ACE2, reduciendo también el potencial de neutralización de sueros convalecientes12-14.
En Bolivia, los últimos reportes indican que se han producido nuevos picos de contagio con variantes de SARSCoV-2. Un estudio genómico realizado por el Instituto Nacional de Laboratorios en Salud (INLASA) detectó en mayo de 2023, la presencia de la variante kraken en Bolivia, más propiamente en el eje troncal de La Paz y Cochabamba (9). Aunque aún no se han reportado estudios de los niveles de IgG-antiRBD en grupos de niños en edad escolar, es muy probable que, debido al bajo número de dosis de vacuna recibidas, estos grupos hayan sido los más vulnerables, tomando en cuenta que las nuevas variantes y sub-variantes como omicron, centaurus y kraken tienen mutaciones que le permiten escapar de la respuesta inmune y además incrementar su afinidad por receptores ACE2.
El uso de herramientas bioinformáticas y el análisis molecular nos permite no solamente describir los procesos relacionados con la mutación de agentes causales de enfermedades como el SARS-CoV-2, sino también, poder predecir si las futuras variantes podrían ocasionar problemas mucho más graves, con el fin de apoyar en la toma de decisiones concernientes a las medidas preventivas y de salud pública.

