









 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Las cromosomopatías son alteraciones en el número o en la estructura de los cromosomas. Las aneuploidías son las Anomalías Cromosómicas (AC) más comunes y de mayor importancia clínica, presentan adición o eliminación de cromosomas dando lugar a trisomías o monosomías1.
Las cromosomopatías producen alteraciones fenotípicas generalmente severas, que incluyen dismorfismos y alteraciones funcionales importantes en diferentes órganos, elevando el riesgo de una muerte prematura. Pueden producirse durante la gametogénesis, por lo que todas las células del sujeto estarán afectadas dando lugar a lo que se conoce como estado libre, o pueden producirse durante la división del cigoto, en la que solo ciertas líneas celulares están alteradas, lo que se conoce como mosaico2,3.
En las cromosomopatías, el fenotipo es muy variado, ocasionando abortos, alteraciones anatómicas leves, malformaciones y disfunciones severas. Sin embargo, hay cierto porcentaje que llega a término, muchos de ellos mueren en el primer año de vida y otros, dependiendo el grado y tipo de lesión que presenten, sobreviven muchos años. En términos generales, las anomalías cromosómicas numéricas autosómicas viables más comunes a nivel mundial son, el Síndrome de Down Libre,4 las alteraciones de cromosomas sexuales como el Síndrome de Turner y el Síndrome de Klinefelter2 son menos frecuentes, y otras cromosomopatías como la trisomía 13 (Síndrome de Patau) o la trisomía 18 (Síndrome de Edward) son raras5.
Por otra parte, las anomalías congénitas son alteraciones del desarrollo fetal, determinadas por diversas causas que actúan antes, durante o después de la gestación6. Los agentes causales pueden ser genéticos, ambientales, por interacción de ambos y en algunos casos por causas desconocidas8. Se caracterizan por presentar alteraciones en la estructura y/o morfología de uno o más órganos y manifestaciones fenotípicas diversas que pueden ser de relevancia clínica importante o insignificante6,7.
El diagnóstico de las alteraciones cromosómicas se realiza con técnicas citogenéticas que van desde el cariotipo convencional, hasta el cariotipo molecular, pasando por las técnicas de hibridación como el FISH y secuenciación. Sin embargo, el cariotipo convencional sigue siendo el gold estándar para el estudio de cromosomas8.
Las malformaciones congénitas en niños menores de un año de edad, en general, son la cuarta causa de muerte en recién nacidos en el mundo y la segunda en las Américas2, siendo que Bolivia está cerca al 3% de mortalidad por malformaciones congénitas teniendo un incremento de 2,5% desde el año 20007.
Las alteraciones cromosómicas se expresan también en el desarrollo psicomotor, el cual es un proceso continuo que va de la concepción a la madurez, con una secuencia similar en todos los niños, pero con un ritmo variable. Mediante este proceso el niño adquiere habilidades en distintas áreas: lenguaje, motora, manipulativa y social, que le permiten una progresiva independencia y adaptación al medio10.
En Cochabamba, muchos recién nacidos con malformaciones o alteraciones funcionales o metabólicas congénitas son diagnosticados solo clínicamente, pues no se realizan los estudios citogenéticos correspondientes; esto contribuye a que muchas enfermedades cromosómicas o alteraciones congénitas que no son clínicamente tan evidentes, pasen desapercibidas. Por ello, las características epidemiológicas de estas alteraciones cromosómicas son desconocidas. El objetivo de este trabajo es describir las características epidemiológicas de las alteraciones cromosómicas y de las malformaciones y disfunciones congénitas en Cochabamba.
Material y métodos
Es un estudio descriptivo transversal en el que se incluyeron 166 pacientes con sospecha de alteración cromosómica en el transcurso de 3 años, referidos del Hospital del Niño Manuel Ascencio Villarroel (centro de tercer nivel), Hospital Rojas Mejía y otros hospitales y centros de salud de la ciudad de Cochabamba, por tanto, los 166 pacientes constituyen la muestra de un universo que comprende toda la población cochabambina. Se incluyeron a todos los pacientes referidos.
A cada paciente se le realizó la anamnesis y exploración física correspondiente previa firma de un consentimiento informado por uno de los padres o apoderados. Se realizó la toma de muestra de sangre periférica y posteriormente se llevó a cabo el estudio citogenético mediante el procedimiento de bandeo cromosómico Giemsa-Tripsina de rutina, adaptado a las condiciones de nuestro laboratorio9. Para la obtención correspondiente del cariotipo se analizaron entre 20 y 40 metafases por cada caso, utilizando el cariotipador MICROPTIC (MetaClass Sistema Cariotipador+FISH). La tabulación y análisis de los datos se realizó mediante software Microsoft Excel y SPSS versión 25.
Resultados
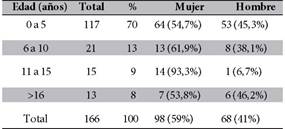
De las 166 personas estudiadas, 98 (59%) fueron Mujeres y 68 (41%) fueron varones. Las edades oscilaron entre recién nacidos y adultos, con una distribución etaria mostrada en la Tabla 1.
De los 166 pacientes estudiados, 79 (48%) tenían cariotipo sin alteración y 87 (52%) tenían alguna anomalía cromosómica. Dentro de los que tenían alguna anomalía, la distribución fue la siguiente: la mayor parte correspondía a un cariotipo de Sd. de Down, (libre y mosaico) (34%), seguido por Sd. de Turner, (11%), Sd. de Edwards, (2%), trisomía 22 (1%), Sd. de Klinefelter (1%), deleciones (2%), cromosoma marcador 5 (1%), (Figura 2).
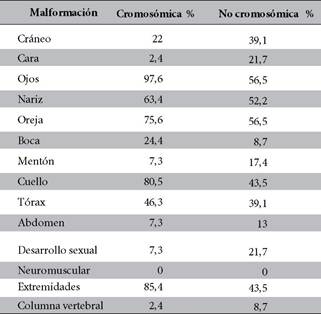

Figura 1. Cariotipo de las principales alteraciones cromosómicas. A. Cariotipo con Sd. Down (Trisomía 21). B. Cariotipo con Sd. Edward (Trisomía 18).
Así mismo, de los 87 pacientes con anomalías cromosómicas, 63 (72,4%) fueron cariotipos libres y 24 (27,6%) fueron mosaicos (Ver Tabla 2). Los diferentes tipos de mosaicismo encontrados para el Sd. de Down fueron: 46,XY,+21/46,XY (n=6) y 46,XX,+21/46,XX (n=2); para el Sd. de Turner se encontraron tres tipos de mosaicismo: 45,X/46,XX (n=8), isocromosoma 45,X/46,X,i(Xq), (n=1) y triple X, 45,X/47,XXX (n=1); el mosaicismo para el Sd. de Edwards: 47,XX,+18/46XX (n=3) (Figura 2); el mosaicismo para el Sd. de Klinefelter: 47,XXY/46,XX (n=1); para la Trisomía 22: 47,XX,+22/46,XX (n=2). Se encontraron también, deleción del brazo largo del cromosoma 18: 46,XY,del(18q); deleción del brazo corto del cromosoma 4: (46,XX,del(4p)); deleción del brazo corto del cromosoma 12: (46,XY,del(12p)), y en un paciente se encontró un marcador.
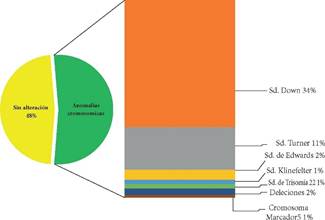
Así mismo, podemos notar que, de acuerdo a la frecuencia de anomalías cromosómicas, la distribución por sexo es (62% mujeres y 38% varones), y es similar a la distribución general (59% mujeres y 41% varones), excepto en el síndrome de Turner que es exclusivo en mujeres.
Malformaciones congénitas en niños menores de un año
De los 166 pacientes atendidos en nuestro laboratorio, 69 (42%) pacientes eran menores de 1 año. La distribución por edad de estos pacientes con dismorfia congènita fue: 10% entre recién nacidos hasta 7 días, 20% neonatos entre 8 y 28 días y 70% lactantes menores y lactantes mayores entre 29 días y un año. De estos, 43 (62%) tenía alteración cromosómica confirmada, y 26 (38%) tenían cariotipo sin alteración (Figura 3). De los 43 pacientes que tenían alteración cromosómica, 81,4% tenían Sd. de Down, 9,3% con Sd. de Edwards (trisomía 18), 2,3% con Sd. de Turner, 2,3% con trisomía 22 y 2,3% con Sd. de Klinefeter. El bajo porcentaje del Sd. de Turner, se debe a que es un síndrome que se diagnostica tardíamente y en este grupo etario solo uno tenía Sd. de Turner.
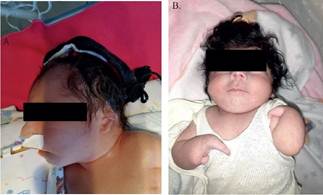
Figura 3. Pacientes atendidos en nuestro laboratorio que presentan alteraciones morfológicas. A. Paciente con Síndrome de Edwards, que presentaba hipoplasia y displasia de oreja, con implantación baja. B. Malformación congénita de miembros superiores (artrogriposis) en una niña con cariotipo sin alteración.
Dismorfias en alteraciones cromosómicas
El análisis de las dismorfias fue hecho en base a una comparación entre los niños con alteración cromosómica y los que tienen cromosomas normales, y nos muestra que las malformaciones congénitas más frecuentes en pacientes con alteración cromosómica fueron: epicanto en ojos en un 97,6%, seguida de cuello corto y dismorfias en extremidades, con 80,5% y 85,4% respectivamente. Las malformaciones en orejas, como implantación baja y microtia entre otras, representan el 75,6%. Las de nariz un 63,4%; las de tórax, 46,3%, dentro de este grupo (Tabla 3).
Desarrollo psicomotor de síndromes cromosómicos
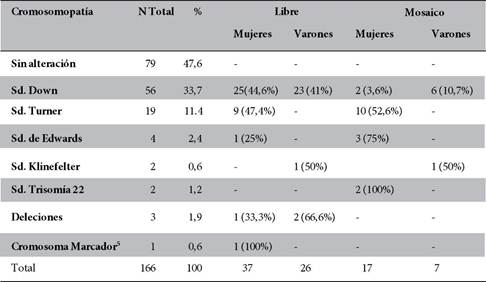
El análisis del retraso psicomotor de los pacientes atendidos, tanto con alteración cromosómica como aquellos sin alteración cromosómica, fue realizada promediando el valor asignado al nivel de desarrollo psicomotor, considerando la edad a la que levantó la cabeza, la edad a la que se sentó y a la que el niño se paró y caminó. Se asignaron valores en una escala de 1 a 5, siendo de 1 a la normalidad, 2 al retraso leve, 3 al retraso moderado 4, al retraso severo, y 5, al retraso muy severo que implicaba inmovilidad total. Este análisis se realizó a tres grupos clasificados según su cromosomopatía: Sd. de Down, Sd. de Turner y sin alteración cromosómica, pero con retraso en el desarrollo.
La distribución de dichos valores de cada síndrome analizado se muestra en la figura 4. Los valores de retraso de los pacientes con Sd. de Down y de los que no tenían alteración cromosómica evidente fueron parecidos, con porcentajes muy cercanos para los distintos grados de retraso; a diferencia de las pacientes con Sd. de Turner que en su mayor parte (90%) no presentan retraso.
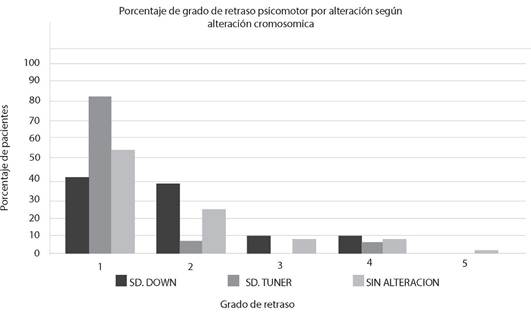
Figura 4. Distribución porcentual de niños con diferente grado de retraso psicomotor según alteración cromosómica. (Valores asignados: 1. Normalidad, 2. Retraso leve, 3. Retraso moderado, 4. Retraso severo y 5. Retraso muy severo, implica inmovilidad total).
Factores de riesgo asociados a cromosomopatías más frecuentes
Siendo la edad de los progenitores de pacientes con diagnóstico de Sd. Down un factor importante. En nuestros datos, de todos los pacientes atendidos, la edad más frecuente de la madre como del padre al tiempo del parto fue entre 21 y 30 años (43,8%, y 41,8% respectivamente), seguida del grupo etario de 31 a 40 años (30,1% y 32,9%) (figura 5A). La edad más frecuente de las madres de pacientes con Sd. Down, fue entre 31 a 40 años (48,1%), seguida del grupo etario de 21 a 30 años (23,1%) y 41 a 50 años (21,2 %), (figura 5C) mientras que la edad de las madres de pacientes con otros síndromes fue de 21 a 30 años (54,5%) y de 31 a 40 años (36,2%) (figura 5B). En el caso de los padres de pacientes con Sd. de Down, la edad más frecuente es de 31 a 40 años (36%), mientras la de los otros pacientes es de 21 a 30 años (51%) y de 31 a 40 años (31,3%).
Para el grupo pacientes con diagnóstico de Sd. de Turner, (Figura 5D), la edad más frecuente de las madres y padres fue de 21 a 30 años (61,1% madres y 55,6 % padres), mientras que para el Sd. de Edwards el porcentaje de casos es igual en todas las edades (datos no mostrados).
Otro factor de riesgo que se destaca, es el número de abortos. Las madres con mayor número de abortos son aquellas que tienen niños con Sd. de Down, sin embargo, la prueba de Chi cuadrado muestra que la asociación entre el número de abortos y el número de madres con hijos con Sd. de Down no es significativa (0,989).
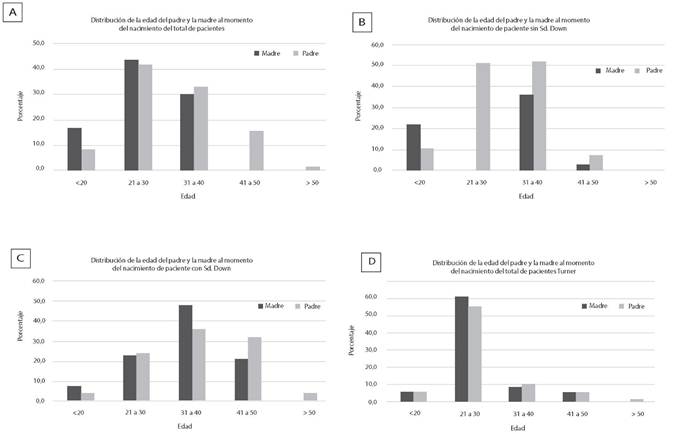
Figura 5. Distribución etaria materna y paterna en los casos de pacietnes con síndromes cromosómicos y no cromosómicos al tiempo de la gestación. En la figura A, se muestra el porcentaje de la edad de madres y padres de todos los pacientes atendidos. En B, distribución de la edad del padre y la madre de pacientes del grupo que no incluye a los pacientes con Sd. Down. En C, distribución de la edad de madres y padres de progenitores de niños con Sd. de Down. En D distribución de la edad de padres y madres de niños con Sd. Turner
Las características socioeconómicas no tienen influencia en la aparición de las alteraciones cromosómicas, y están descritas en los siguientes Figuras (Figura 6).
Respecto a la escolaridad de los padres de los pacientes atendidos, encontramos que ésta fue más frecuentemente de nivel bachiller o universitario, es decir que la mayor parte de los pacientes provenían de un ambiente familiar con grado de instrucción media y profesional (Figura 6).
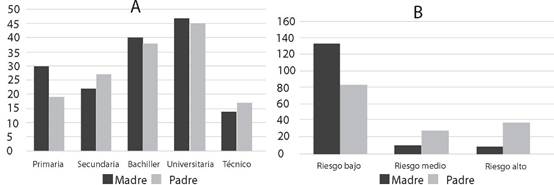
Figura 6. A) Grado de escolaridad de los progenitores de pacientes atendidos. B) El riesgo ocupacional de enfermedades genéticas fue calculado a partir de la actividad de los padres.
Otro factor de riesgo genético es la cercanía a algún elemento que pudiera afectar la concepción o gestación, lo que se puede presentar en el tipo de actividad económica que realizan los padres. En este estudio, calculamos este riesgo a partir de la evaluación del entorno de la actividad económica en la que se desempeñaba la madre o el padre. Fueron calificados como de riesgo alto actividades como: albañil, tornero, artesano, herrero, pintor, laboratorista, vendedor de sustancias químicas para el padre, y agricultora, limpieza, laboratoristas para la madre.
La gráfica 6B muestra que en general el riesgo es bajo y medio, pero de los que fueron clasificados como riesgo alto, los padres están en mayor frecuencia que las madres.
El área de residencia de los pacientes atendidos es un parámetro importante para conocer posibles riesgos externos. La distribución que encontramos fue de 40% provenían de zonas periféricas, 37% del área rural y 23 % del área urbana.
Discusión
Las anomalías cromosómicas (AC) son una de las principales causas de discapacidad infantil, en especial las aneuploidías11. En nuestro estudio encontramos que, del total de pacientes con sospecha de anomalías cromosómicas (AC) que acudieron a realizarse la prueba de cariotipo, prácticamente la mitad no presentó AC ; pero dentro del grupo de pacientes que sí tuvieron, las aneuploidías más frecuentes fueron el Sd. Down, seguido del Sd. Turner y Trisomía 18. Estos resultados son similares a los reportados en la literatura12,13. Cabe recalcar que el sexo femenino fue el grupo donde las AC están en mayor frecuencia (60%) a pesar de que otros estudios reportan lo contrario. Sin embargo, el sexo no es considerado un factor predisponente para las AC, excepto en el síndrome de Turner que es exclusivo en mujeres (12). Los cariotipos encontrados para Sd. Down y los otros síndromes fueron libres en la mayor parte, excepto para el Sd. de Turner para el que encontramos cerca al 50 % de los pacientes con distintos mosaicos, como se reporta también en la literatura14.
Encontramos también que el porcentaje de niños menores de 1 año con malformaciones congénitas sin alteración cromosómica es de solo 32%, lo cual nos muestra que la mayor proporción de alteraciones congénitas se debió a alteración cromosómica.
En cuanto al retraso del desarrollo psicomotor, en nuestro estudio, éste puede ser atribuido, en su mayor parte a cromosomopatías como Sd. Down y Sd. de Edward. Cabe recalcar que los retrasos en el desarrollo motor como de lenguaje, pueden ser temporales y no estar sujetos a un pronóstico de retraso mental u otra discapacidad cognitiva15. Haciendo una comparación del retraso del desarrollo psicomotor entre el Sd. Down, Sd. de Turner y los que no tenían evidencia de alteración cromosómica, nuestros datos muestran que el Sd, Down afecta en un mayor porcentaje, mientras que los de Sd de Turner son muy poco afectados en esta área. Los que no tenían alteración cromosómica fueron también afectados, pero en menor grado que los con Sd. de Down.
Uno de los factores de riesgo de las cromosomopatías considerado como más frecuente en el Sd. Down, es la edad de los progenitores. En nuestro estudio la frecuencia de padres de niños con Sd. Down fue aumentando conforme la edad de la madre y del padre era mayor, lo que implica que existe un factor en los progenitores que induce a la trisomia, no obstante, también se presentó en padres con edades tan jovenes como menores de 20 años y entre 20 y 30, lo que muestra que la edad de la madre fue un factor asociado, pero no exclusivo por lo que, la mutación puede deberse a otros factores no relacionados a la edad como también lo refiere M. Vashist y Neelkamal16. Por otra parte, es interesante que la edad de las madres de niños con Sd. de Turner, sea más bien joven. Si la edad es un factor de riesgo, se espera que la distribución de hijos con Sd. de Turner sea uniforme, sin embargo, la frecuencia mayor fue de padres entre 20 a 30 años, lo que sugiere pueda haber un factor no asociado a la edad, sino al ambiente, como, por ejemplo, el ambiente de la gestación (post concepción), que podrían estar presentes en estas madres, ya que la mitad de ellas tuvo aborto, es decir, un factor adicional. La edad del padre también es importante, ya que en nuestro estudio, la mayor parte de los casos eran hijas de padres menores de 30 años, lo que podría estar relacionado a los reportes de que la mayor frecuencia de la mutación es de origen paterno17. En los otros síndromes, la edad de los padres no parece influir.
Respecto a los factores socioeconómicos y la escolaridad, ninguno de los dos parece ser un factor de riesgo, ya que la mayor parte tiene, según nuestra clasificación, riesgo bajo.
En conclusión, encontramos que la prevalencia de las alteraciones cromosómicas en Cochabamba es similar a la reportada en otros países. Las aneuploidías más frecuentes en Cochabamba fueron el Sd. Down, seguido del Sd. Turner y Trisomía 18. Los cariotipos de Sd. Down y otros fueron libres en la mayor parte, excepto para el síndrome de Turner que tiene mosaicos en alto porcentaje. La mayoría de los dimorfismos se deben a la cromosomopatía. La edad de la madre y del padre parece ser un factor de riesgo para el Sd. Down.