










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkGaceta Médica Boliviana
On-line version ISSN 1012-2966
Gac Med Bol vol.43 no.2 Cochabamba Dec. 2020
Artículos de Revisión
SARS-CoV-2: estructura, replicación y mecanismos fisiopatológicos relacionados con COVID-19
SARS-CoV-2: structure, replication and physiopathological mechanisms related to COVID -19
Arandia-Guzmán Jaime1, Antezana-Llaveta Gabriela2
Recibido el 07 de julio de 2020
Aceptado el 16 de septiembre de 2020
Resumen
SARS-CoV2 es causante del síndrome respiratorio agudo severo, enfermedad que ha sido también nombrada COVID-19, fue notificado a fines del año 2019 como un nuevo betacoronavirus en personas expuestas en un mercado de mariscos en Wuhan, China. El virus desde esa fecha se ha ido propagando rápidamente provocando una pandemia, y afectando a diversos países en mayor o menor magnitud. Actualmente existe información variada difundida sobre el virus y la enfermedad; los conocimientos sobre la fisiopatología y la manera en la que debe ser gestionada esta entidad han ido cambiando a través de tiempo. A pesar del interés que se ha generado en los mecanismos fisiopatológicos de la enfermedad y sus complicaciones, estos no se han llegado a descifrar totalmente. Mediante el presente artículo se hace una revisión sistematizada de la estructura, replicación y aspectos fisiopatológicos relacionados con SARS-CoV2, que ha provocado un elevado índice de morbimortalidad en la población a nivel mundial.
Palabras clave: SARS-CoV-2, COVID-19, betacoronavirus, mecanismos fisiopatológicos, morbimortalidad.
Abstract
SARS-CoV2, which causes severe acute respiratory syndrome, also called COVID-19, was reported in late 2019 as a new betacoronavirus in exposed persons in a seafood market in Wuhan, China. The virus has since spread rapidly, causing a pandemic, affecting several countries to a greater or lesser extent. Currently, there is a variety of information disseminated about the virus and the disease; knowledge about the physiopathology and how it should be managed has changed over time. Despite the interest that has been generated in the physiopathological mechanisms of the disease and its complications, these have not been completely deciphered. The present article makes a systematized review of the structure, replication and physiopathological aspects related to SARS-CoV2, which has caused a high rate of morbimortality in the population worldwide.
Keywords: SARS-CoV-2, COVID-19, betacoronavirus, physiopathological mechanisms, morbidity.
El nuevo coronavirus del síndrome respiratorio agudo severo (SARS-CoV2) es un virus que pertenece a la familia Coronaviridae, subfamilia Coronavirinae1. Es causante del síndrome respiratorio agudo severo o también llamado COVID-19 que fue notificado a fines del 2019 como un nuevo betacoronavirusen personas expuestas en un mercado de mariscos en Wuhan, China1,2. Los brotes del síndrome respiratorio agudo severo (SRAS) en 2002/2003 y el síndrome respiratorio del Medio Oriente (MERS) en 2012, también causados por especies de coronavirus han demostrado la posibilidad de transmisión de animales a humanos y de humanos a humanos2,3. SARS-CoV-2 comparte alrededor de 96,2% aproximadamente de homología de secuencia con coronavirus de murciélago y actualmente, se cree que este virus ha sido introducido en humanos por un animal intermediario aún no identificado y luego se ha propagado de humano a humano3.
Los síntomas de COVID-19, enfermedad nombrada de esta manera por el Dr. Tedros Adhanom Ghebreyesus, Director General de la Organización Mundial de la Salud (OMS) el 11 de febrero de 2020, varían dependiendo de si se trata de una forma leve, moderada o severa de la enfermedad4. En casos de COVID-19 leve los síntomas pueden incluir alzas térmicas, tos seca, malestar general, mialgias, anosmia y ageusia; algunos pacientes tienen síntomas gastrointestinales, como anorexia, naúseas, vómitos y diarrea1. Los casos graves de COVID-19 se producen sobre todo en pacientes con enfermedad crónica de base como ser patología cardiovascular, diabetes mellitus, enfermedad renal crónica y obesidad entre otros; sin embargo también se han reportado en pacientes sin comorbilidad de cualquier edad5,6. Estos pacientes pueden tener complicaciones graves como el síndrome de distrés respiratorio agudo (SDRA) con disnea e hipoxemia, linfopenia, también pueden existir trastornos del sistema nervioso central o periférico, falla renal, insuficiencia cardiaca, falla hepática, cuagulopatías, y shock5-8.
Con el presente artículo se tratará de dilucidar las bases moleculares de la fisiopatología del COVID-19, que nos ayudará a comprender no sólo la enfermedad, sino también sus complicaciones y los mecanismos necesarios para llegar a la falla multiorgánica que se produce en varios casos reportados de esta enfermedad (Ver figura 1).
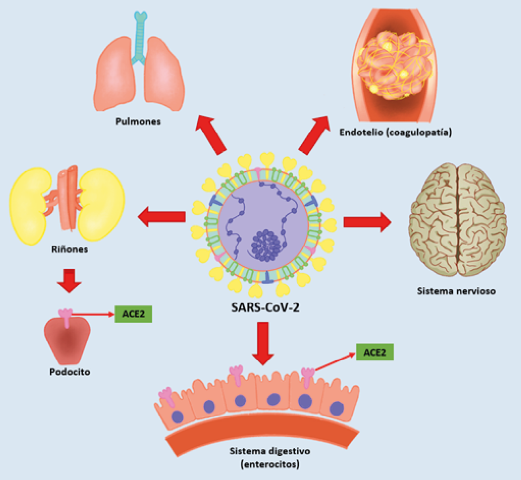
ser afectados por SARS-CoV-2, debido a la presencia de receptores de la enzima convertidora de angiotensina 2
(ACE2) en distintos órganos, incluyendo pulmones, riñones, neuronas y enterocitos entre otros.
Fuente: elaboración propia.
Revisión bibliográfica
Para el presente artículo se realizó una búsqueda sistemática exhaustiva de fuentes primarias de información en buscadores online e indizaciones científicas, como ser: PubMed, ElSevier, Scielo, MedLine, etc.
Las fuentes primarias propiamente dichas corresponden a artículos científicos publicados y pre publicados (Review Articule, Guías Clínicas, Case Report, Serie de Casos y Ensayos Clínicos) que serán mencionados en la bibliografía final.
Desarrollo y discusión del tema
Estructura general de SARS-CoV-2
Se reconocen cuatro géneros de coronavirus: Alphacoronavirus, Betacoronavirus, Gammacoronavirus y Deltacoronavirus2. El exámen genealógico del SARS-CoV-2 reveló que pertenece al coronavirus del género betacoronavirus4.
Los coronavirus reciben su nombre debido al aspecto que presentan sus viriones al microscopio electrónico, semejante a una corona solar (con proyecciones de superficie) gracias a sus proteínas de superficie9. Estructuralmente los coronavirus son virus esféricos que miden entre 80 a 160 nanómetros de diámetro, con una envoltura de bicapa lipídica y que contienen genoma de ARN monocatenario (ssRNA) de polaridad positiva de entre 27 y 30 kilobases de longitud9. El genoma del virus SARS-CoV-2 codifica 5 proteínas estructurales, las cuales están codificadas dentro del extremo 3’ del genoma viral9, 10, 11:
Glucoproteína S (espiga): La glucoproteína S trimérica es una proteína de fusión de clase I y media la unión al receptor del huésped. La glucoproteína S es escindido por una proteasa similar a la furina de la célula huésped en dos polipéptidos separados denominados S1 y S2. S1 constituye el gran dominio de unión al receptor de la proteína S, mientras que S2 forma el tallo de la molécula espiga.
Proteína E (envoltura): La proteína E transmembranal tiene un ectodominio N-terminal y un endodominio C-terminal y tiene actividad de canal iónico. La actividad del canal iónico en la proteína E del SARS-CoV no es necesaria para la replicación viral, pero sí podría serlo para la patogénesis. Facilita el ensamblaje y la liberación del virus.
· Proteína N (nucleocápside): Se compone de dos dominios separados, un dominio N-terminal y un dominio C-terminal, ambos capaces de unirse al ARN in vitro, pero cada dominio utiliza diferentes
mecanismos para unirse al ARN. La proteína N también está muy fosforilada, y se ha sugerido que la fosforilación desencadena un cambio estructural que mejora la afinidad por el ARN viral versus el no viral. La proteína N se une al genoma viral en una conformación de tipo perlas en una cuerda.
· Hemaglutinina-esterasa (HE): Está presente en un subconjunto de betacoronavirus. La proteína actúa como una hemaglutinina, se une a los ácidos siálicos en las glucoproteínas de superficie y contiene actividad acetil-esterasa. Se cree que estas actividades mejoran la entrada de células mediadas por la proteína S y la propagación del virus a través de la mucosa.
Entre estas cinco proteínas, las más importantes son la proteína N y la proteína S, donde la primera ayuda al virus a desarrollar la cápside y la estructura viral completa de manera apropiada y la última ayuda a la unión del virus a las células del huésped11 (Ver figura 2).
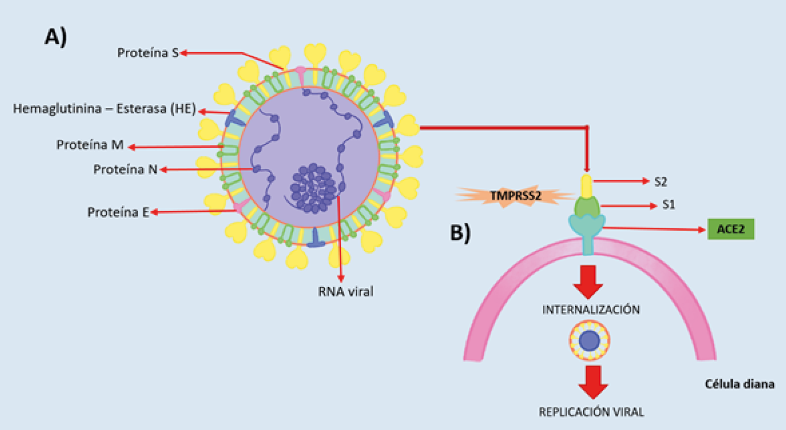
Figura 2. Estructura de SARS-CoV-2, virus causante de COVID-19 con sus proteínas estructurales correspondientes(A). Mecanismo de
unión de la proteína S al receptor de la enzima convertidora de angiotensina 2 (ACE2). En la figura se puede apreciar como la proteína S
es escindida por la serina proteasa TMPRRS2, en la subunidad S1 (N- terminal) y S2 (C- terminal) mediando la unión del virus a la célula
diana y facilitando el ingreso del mismo13,14 (B).
Fuente: elaboración propia.
Se ha informado que SARS-CoV-1(agente causal del síndrome respiratorio agudo severo en 2002/2003) y SARS-CoV-2 (agente causal de COVID-19) tienen un tipo similar de receptores. La proteína S se une directamente al receptor de la enzima convertidora de angiotensina 2 (ACE2) en las células diana del huésped10,11. El receptor de ACE2 se expresa en varios órganos del cuerpo humano, principalmente en los pulmones, los riñones y el intestino que representan los principales objetivos del coronavirus11, 12. La afinidad de unión del SARS-CoV-2 al receptor ACE2 es de 10 a 20 veces mayor en comparación con el SARS-CoV-112.
Se ha encontrado que los virus más patógenos contienen un sitio de escisión similar a la furina en la proteína S, que no está presente en el SARS-CoV-1 pero sí en el SARS-CoV-2; este proceso requiere serinas proteasas celulares (TMPRSS2), que permiten la escisión de la proteína S, regulando todo el mecanismo y mejorando así la fusión viral con las membranas de las células huésped13. La proteína S posee así dos subunidades funcionales S1 (N-terminal) y S2 (C-terminal) mencionadas anteriormente; el primero media la unión del virus a la membrana de la célula huésped reconociendo un receptor en la célula afín , mientras que el segundo favorece la fusión de las 2 membranas celulares, y está implicada en la entrada viral14.
Replicación de SARS-CoV-2
Una vez que el virus ha ingresado a la célula huésped, este inicia el proceso de replicación; el genoma del virus contiene un gran gen replicasa que dará lugar a proteínas no estructurales (Nsps), seguido de genes estructurales y accesorios9,15. El gen replicasa codifica dos marcos de lectura abiertos (ORF), rep1a y rep1b, que se traducen en dos poliproteínas (pp1a y pp1ab); estos polipéptidos son procesados por dos proteasas virales: la proteasa tipo 3C (3CLpro) y la proteasa tipo papaína. La escisión produce 15 o 16 Nsps virales que se ensamblan en un gran complejo unido a membrana y exhiben múltiples actividades enzimáticas15.
El genoma de ARN de cadena positiva se usa como plantilla para producir la cadena negativa. Las enzimas codificadas por el gen replicasa usan el ARN negativo como plantilla para desarrollar segmentos de ARN mensajero (ARNm) superpuestos que se traducen en las proteínas estructurales11. Se cree que la fabricación de estas moléculas individuales de ARN podría favorecer sucesos de recombinación entre genomas víricos y diversidad genética9.
Durante el proceso de replicación dentro del huésped humano, la proteína N del virus se une al genoma, mientras que la proteína M se asocia con las membranas del retículo endoplásmico (RE). Posteriormente el ARN mensajero y las proteínas de nucleocápside se combinan para formar los viriones11. Las partículas virales se dirigen al complejo intermediario retículo endoplásmico - aparato de Golgi y desde este compartimiento las vesículas que contienen los viriones se dirigen a fusionarse con la membrana plasmática, armando así las partículas virales completas que al ser liberadas pasan a infectar nuevas células16.
El primero está mediado por la transcripción de los interferones tipo I y III (IFN-I e IFN-III, respectivamente) y la posterior regulación positiva de los genes estimulados por IFN. El segundo implica el reclutamiento y la coordinación de subconjuntos específicos de leucocitos, que se caracteriza por la secreción de quimiocinas19,21. Se ha postulado que la respuesta del huésped al SARS-CoV-2 no puede lanzar una respuesta robusta de IFN-I y III al tiempo que induce altos niveles de quimiocinas necesarias para reclutar células efectoras19,22.
Las citocinas y las quimiocinas proinflamatorias que se elevaron durante la infección con COVID-19 incluyen el factor de necrosis tumoral α (TNF- α), interleucina 1β (IL-1β), IL-6, factor estimulante de colonias de granulocitos (G-CSF), proteína 10 inducida por interferón gamma (IP-10), proteína quimioatrayente de monocitos 1 (MCP-1) y las proteínas inflamatorias de macrófagos 1- α (MIP 1- α)23.
SARS-CoV-2 ingresa a los neumocitos tipo 2 en humanos a través del receptor ACE2, después inicia el proceso de replicación descrito anteriormente13. Xu y col. realizaron estudios histopatológicos con muestras de biopsia de tejido pulmonar en pacientes con COVID-19 y han demostrado la presencia de daño alveolar difuso con la formación de membranas hialinas, presencia de células mononucleares y macrófagos infiltrando espacios aéreos con un engrosamiento difuso de la pared alveolar24. También se han observado partículas virales en neumocitos tipo II mediante microscopía electrónica23. Estas alteraciones patológicas descritas pueden llevar al paciente a un síndrome de dificultad respiratoria aguda severa (SDRA), caracterizado por hipoxemia severa, aparición aguda de infiltrado bilateral que ha sido descrito en patrón de “vidrio esmerilado” en las placas radiográficas, y edema pulmonar que no se explica por sobrecarga de líquidos ni insuficiencia cardiaca5.
En la patogénesis del SDRA existe una lesión epitelial con permeabilidad aumentada en la membrana alveolocapilar, además de un daño difuso de las células alveolares que da lugar a la acumulación de líquido, inactivación del tensioactivo o surfactante que es producido por los neumocitos tipo 2 y posteriormente formación de la membrana hialina que se considera impermeable al intercambio de gases; estos cambios que ocurren a nivel pulmonar ocasionarían un incremento del trabajo respiratorio en el paciente con signos de dificultad respiratoria, además existiría una derivación intrapulmonar con incremento de flujo sanguíneo que también afectaría el intercambio de gases con la aparición de hipoxemia refractaria en estos pacientes25.
SARS-CoV-2 y coagulopatía
La coagulopatía es una característica común de la infección por SARS-CoV-2, suele estar caracterizada por incremento de dímero D y fibrina como hallazgo común en la analítica26.
El dímero D es el producto final de la degradación de fibrina que sirve como indicador serológico de la activación de la coagulación y del sistema fibrinolítico27.
En pacientes con COVID-19 grave se ha descrito que existe una lesión local directa vascular y endotelial que produce formación de coágulos microvasculares y angiopatía; también existe un estado de hipercoagulabilidad con hiperfibrinogenemia y la posibilidad de trombosis de grandes vasos o secuelas tromboembólicas importantes, incluida la tromboembolia pulmonar (TEP), que se reporta hasta en el 20 a 30% de los pacientes de unidad de cuidados intensivos (UCI)28.
La cascada de coagulación se activa en infecciones virales como es el caso de COVID-19; existe un desequilibrio entre los mecanismos homeostáticos procoagulantes y anticoagulantes en esta patología; como resultado del aumento de la actividad inflamatoria los niveles de fibrinógeno aumentan y se forman los trombos29,30 (Figura 3).
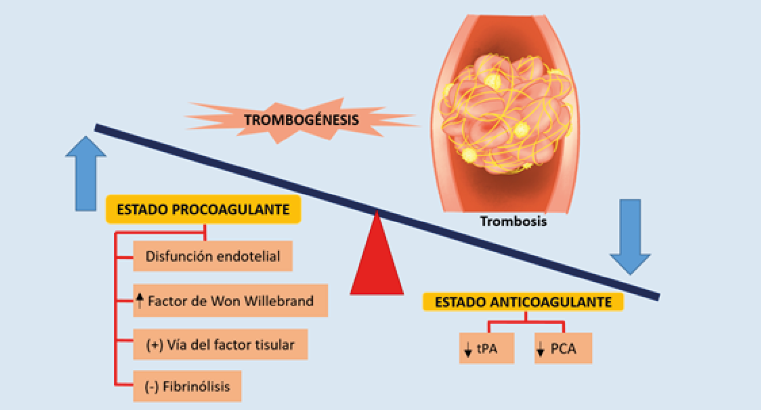
Figura 3. Fisiopatología de la coagulopatía en pacientes con COVID-19. En la figura se observa una desregulación de los mecanismos
de coagulación normal, con un estado procoagulante que favorece que la formación de trombos y complicaciones tromboembólicas.
Existe mayor expresión de factores que favorecen a la coagulación como el factor de Von Willebrand, activación de la vía del factor
tisular, inhibición de la fibrinólisis y disfunción endotelial; por otro lado existe menor expresión de factores anticoagulantes como el
activador tisular de plasminógeno (tPA) y la proteína C (PCA)28,30.
Fuente: elaboración propia.
El aumento de los niveles de productos de degradación de fibrina como el díametro D y el tiempo prolongado de protrombina se han asociado con un mal pronóstico en pacientes afectados por el nuevo betacoronavirus y múltiples mecanismos patogénicos se han involucrado en la coagulopatía ocasionada por COVID-19, incluyendo disfunción endotelial, elevación del factor Von Willebrand, activación del receptor tipo Toll y activación de la vía del factor tisular30.
La asociación con el aumento de la trombosis microvascular, del aumento de los niveles de lactato deshidrogenasa (LDH) y ferritina, y los aumentos leves en tiempo de protrombina (PT) y tiempo de trombopastina parcial activada (aPTT) están en relación con una microangiopatía trombótica31.
La prevalencia de trombosis arterial también es elevada en paciente con COVID-19 en quienes se ha sugerido la participación de anticuerpos antifosfolípidicos30.
En un estudio se examinó la presencia de trombosis en miembros inferiores sin síntomas de trombosis venosa profunda (TVP) mediante ultrasonografía en pacientes con neumonía por COVID-19 tratados en unidad de cuidados intensivos (UCI) e informaron que la prevalencia era del 25%; este hecho podría estar favorecido por el tiempo que permanecen estos paciente en cama, por el estado procoagulante descrito que ocurre en pacientes con COVID-19 y también podría deberse a un estado de inhibición fibrinolítico32. La presencia de trombosis venosa profunda (TVP) es un factor de riesgo reconocido para presentar TEP.
Debido a la coagulopatía existente en paciente con COVID-19, actualmente existen guías que recomiendan el uso de la anticoagulación en pacientes con COVID-19; incluso existen estudios que muestran una mortalidad reducida en casos con coagulopatía y tratados con heparina (heparina de bajo peso molecular) en comparación con los pacientes que sí tenían coagulopatía y no fueron tratados con heparina33,34. Sin embargo el tratamiento en relación a las alteraciones de la coagulación que se producen en esta enfermedad va más allá del alcance de este artículo.
La injuria renal aguda (IRA) representa una complicación grave en pacientes críticos, que a menudo conduce a un mayor riesgo de muerte37.
Entre los posibles mecanismos patogénicos del SDRA asociado con IRA se describen las alteraciones hemodinámicas, incluida la insuficiencia cardíaca derecha, la sobrecarga de líquidos y la congestión sistémica (incluyendo congestión de la vasculatura renal), las estrategias de ventilación mecánica perjudicial, el desarrollo de infecciones secundarias, la reacción inflamatoria inmunomediada con la liberación de altos niveles de mediadores inflamatorios circulantes dañinos que son capaces de interactuar con las células residentes en los riñones y de causar disfunción endotelial, trastorno microcirculatorio y lesión tubular; también se ha postulado una lesión renal mediada por el propio virus en glomérulo y en sistema tubular36,38.
Un estudio realizado por Pan y col. mostró una coexpresión relativamente alta de receptor ACE2 y de la serina proteasa TMPRSS en los podocitos y las células del túbulo recto proximal del glomérulo renal, que se identificaron como células huésped candidatas en IRA38.
De esta manera se ha sugerido una IRA prerrenal, pero también se pueden encontrar datos de glomerulonefritis e incluso ciertas formas de necrosis tubular aguda, que sugieren componentes de una IRA intrarenal en estos pacientes39. En la clínica, los pacientes pueden cursar con proteinuria (probablemente por el daño podocitario), hematuria, oliguria y un incremento de los niveles de creatinina sérica36. Se han descrito en otro estudio realizado por Hirsch y col. los posibles factores predictores de IRA en COVID-19 citando entre ellos: la edad avanzada, el sexo masculino, la diabetes mellitus, la hipertensión arterial, antecedente de enfermedad cardiovascular, aumento del índice de masa corporal (IMC), la ventilación mecánica, los medicamentos vasopresores y un historial de tratamiento con medicamentos inhibidores de la angiotensina-aldosterona renal39.
Recientemente de igual manera se ha descrito un caso de COVID-19 asociado a glomerulopatía colapsante que corresponde a la variante más temida de glomerulonefritis focal y segmentaria, esta patología comúnmente se ha asociado con infecciones virales como el virus de la inmunodeficiencia humana, parvovirus, citomegalovirus, enfermedades autoinmunes, el consumo de heroína y mayor predisposición en sujetos de raza negra40.
La diarrea es un síntoma frecuente en las infecciones por coronavirus; se ha detectado hasta en el 30% de los pacientes con MERS y el 10,6% de los pacientes con SARS-CoV-1941.
El receptor ACE2 necesario para la entrada del virus también se expresó en células de esófago superior así como en enterocitos de íleon y colon; TMPRSS2 también se encuentra coexpresado a nivel de los enterocitos y en el esófago, lo cual explicaría la presencia de manifestaciones gastrointestinales en paciente con COVID-1912. En la patogénesis del síndrome diarreico producido por SARS-CoV-2 se postula que la infección viral causa una alteración de la permeabilidad intestinal, lo que resulta en una mala absorción por parte de los enterocitos42. El receptor ACE2 es necesario para la expresión superficial de transportadores de aminoácidos del intestino delgado y se ha sugerido que la actividad del virus puede causar modificaciones enzimáticas, lo que aumenta la susceptibilidad a la inflamación intestinal y la diarrea; ya que aminoácidos como el triptófano regulan la secreción de péptidos antimicrobianos por las células de Paneth a través de la activación de la vía mTOR, estos péptidos antimicrobianos podrían afectar la composición y diversidad de la microbiota contribuyendo a la patogenia provocando enteritis (inflamación del intestino delgado) y en última instancia diarrea12.
Se han descrito manifestaciones neurológicas características en algunos pacientes con COVID-19, lo que indica que el SARS-CoV-2 podría representar un patógeno oportunista subestimado del cerebro43.
De hecho se han utilizado varias técnicas, como la inmunohistoquímica, la microscopía electrónica y técnica de reacción en cadena de la polimerasa (PCR) y se ha detectado la presencia de coronavirus en el cerebro44,45.
Se postulan como posibles rutas de ingreso de SARS-CoV-2 hacia el sistema nervioso central (SNC): la transferencia transináptica en neuronas infectadas, a través del nervio olfatorio, la vía linfática y a través la barrera hematoencefálica46.
Se cree que existe un transporte axonal retrógrado de SARS-CoV-2, donde se utilizan los microtúbulos axonales para facilitar el transporte del virus hacia el SNC; la ruta de ingreso en este caso se piensa que son las neuronas olfatorias, ya que se ha descrito un estudio con ratones en los cuales la ablación química de estas neuronas los protegía de la infección47 (Figura 4).
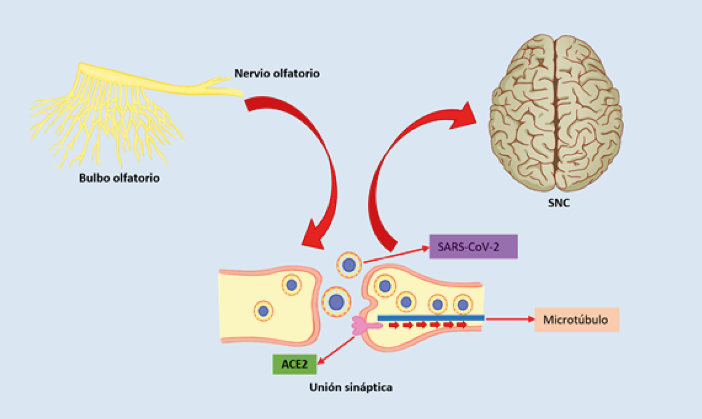
Figura 4. Posible ruta de ingreso de SARS-CoV-2 hacia el Sistema Nervioso Central (SNC), ingresando a través de las neuronas olfatorias,
utilizando un transporte retrógrado neuronal que es mediado por microtúbulos47. Fuente: elaboración propia.
En el caso de que SARS-CoV-2 ingrese al SNC atravesando la barrera hematoencefálica (BHE) podría ser gracias a una infección previa de la células endoteliales que forman parte de esta barrera (junto con los astrocitos, pericitos y matriz extracelular) debido a la gran cantidad de receptor ACE2 que contienen estas células; una vez que ha llegado a este punto el virus sería capaz de invadir el tejido adyacente y cruzar la BHE ocasionando un daño vascular y neuronal47. Los leucocitos también podrían llevar el virus a través de la sangre hacia la BHE, este mecanismo de transporte hacia el SNC estaría facilitado por un aumento de la permeabilidad de la BHE que ocurre en la inflamación ocasionada por la infección de SARS-CoV-2 facilitando así el paso del mismo46,48.
Los síntomas neurológicos más comunes que se han descrito son cefalea, anosmia y ageusia y como otros hallazgos neurológicos se incluyen accidente cerebrovascular (ACV), deterioro de la conciencia, coma, convulsiones y encefalopatía46. Toscano y col. reportaron casos de COVID-19 asociados a síndrome de Guillain-Barré (SGB), que presentaron de 5 a 10 días posterior a la infección, el SGB podría explicarse por un mimetismo molecular en el que los virus infecciosos compartirían epítopos similares a los componentes de los nervios periféricos, lo que estimula las células T o B autorreactivas, un mecanismo similar al que ocurre con las infecciones por Campylobacter Jejuni y algunas infecciones virales como por el virus de Epstein-Barr, citomegalovirus y virus del Zika49.
Las manifestaciones cutáneas asociadas a COVID-19 han sido reportadas en algunas series de casos. Se han descrito patrones desde eritema maculopapular, urticaria, lesiones vesiculares, púrpura periflexural, livedo reticularis transitorio, acroisquemia, hasta eritema multiforme50,51. Los mecanismos fisiopatológicos de las alteraciones cutáneas de COVID-19 aún no se conocen bien y todavía no está claro si las alteraciones cutáneas son secundarias de la infección respiratoria o son por el contrario una infección primaria de la piel misma; se postula que las partículas virales presentes en los vasos sanguíneos cutáneos en pacientes con infección por COVID-19 podrían conducir a una vasculitis linfocítica ocasionando estas alteraciones52. También se describe este hallazgo relacionado con una diseminación hematógena del virus a través del sistema vascular cutáneo donde también existiría una activación del sistema inmune con la movilización de linfocitos y células de Langerhans; las partículas virales podrían inducir la creación de complejos inmunes, esto llevaría a los linfocitos T cooperadores (CD4) a producir citocinas, como IL-1, IFN-γ y TNF-α y a reclutar eosinófilos, células T citotóxicas (CD8), células B y células asesinas naturales (NK) que provocan una arteritis trombofílica linfocítica53.
Actualmente si bien existen series de casos reportados y artículos que se refieren a las lesiones dérmicas asociadas a COVID-19, se necesitan más estudios para evaluar si estas lesiones están asociadas con el virus de manera directa, o es atribuible a la respuesta inflamatoria ocasionada por el virus, y los detalles de los mecanismos patológicos involucrados.
Babapoor-Farrokhran y col. han sugerido que la enfermedad cardiovascular subyacente puede predisponer a los pacientes a un mayor riesgo de infección por coronavirus a través del aumento de la inflamación sistémica y la desregulación del sistema inmunológico; sin embargo no hay evidencia científica que respalde esta afirmación54.
Actualmente existe una creciente literatura que explora las lesiones cardiacas que podrían estar asociadas a COVID-19. Se sabe que el receptor de ACE2 se expresa en el tejido cardiaco específicamente a nivel de los pericitos, paralelamente en un estudio realizado por Burrel y col. se ha visto que el receptor ACE2 esta expresado en mayor magnitud en pacientes con patología cardiaca subyacente en relación a personas con tejido cardiaco normal55,56.
Los pacientes que reciben fármacos como antagonistas del receptor de angiotensina (ARA) o inhibidores de la enzima convertidora de angiotensina II (IECA) tienen una regulación positiva de la ACE2, por lo tanto este receptor estaría disponible en grandes cantidades para ofrecer un sitio
de unión para el SARS-CoV-257. Las manifestaciones cardíacas de COVID-19 se han relacionado con una respuesta adrenérgica, el estado inflamatorio sistémico, infección viral directa de células endoteliales y miocárdicas, hipoxia debida a insuficiencia respiratoria, desequilibrios electrolíticos, sobrecarga de líquidos y efectos secundarios de ciertos medicamentos dirigidos contra COVID-1957,58,59.
Zou y col. en un estudio de metanálisis examinaron la
Las arritmias cardiacas en estos pacientes han sido atribuidas a diversos factores, entre ellos la inflamación del miocardio provocada por una infección viral que conduce a un remodelado electrofisiológico y estructural; pero también las arritmias pueden ser secundarias a efectos secundarios de la medicación; la fibrilación auricular fue la arritmia cardíaca más observada en pacientes con infección por COVID-1961. Existen reportes de arritmias ventriculares y torsada de pointes como complicaciones de efectos adversos debido a medicamentos que prolongan el intervalo QT en el electrocardiograma, especialmente azitromicina e hidroxicloroquina61,62.
Los pacientes con antecedentes médicos de enfermedad coronaria son propensos cardiopatía inducida por COVID-19, además los pacientes pueden desarrollar síndrome coronario agudo sobre todo como complicación trombótica63.
Conclusiones
SARS-CoV-2 corresponde a un nuevo betacoronavirus que se considera un virus emergente en la población mundial. Conocer la estructura y replicación del virus resulta esencial para conocer los aspectos patológicos del mismo. Hasta la fecha, el SARS-CoV-2 ha infectado a millones de personas y ha afectado miles de millones de vidas. La comprensión de la enfermedad y su fisiopatología en pacientes con COVID-19 está evolucionando, y los médicos deben continuar realizando investigaciones, para comprender no solo los efectos que tiene la infección en los diferentes tejidos y órganos, sino también para conocer las posibles complicaciones que esta enfermedad podría tener a corto y a largo plazo, cuyo conocimiento también podría facilitar el manejo y tratamiento de estos pacientes.
Referencias bibliográficas
1. Gandhi RT, Lynch JB, Del Rio C. Mild or moderate Covid-19. N Engl J Med. 2020 April 24. [ Links ] DOI: 10.1056/NEJMcp2009249 [E-pub ahead of print]. https://doi.org/10.1056/NEJMcp2009249
2. Chen Y, Liu Q, Guo D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J Med Virol. 2020; 92: 418-23. [ Links ] https://doi.org/10.1002/jmv.26234
3. Cui J, Li F , Shi Z L. Origin and evolution of pathogenic coronaviruses. Microbiology. 2019; 17(3): 81-192. [ Links ] https://doi.org/10.1038/s41579-018-0118-9
4. Renu K, Prasanna P L , Gopalakrishnan A V. Coronaviruses pathogenesis, comorbidities and multi-organ damage - A review, Life Sci. 2020 May 22. [ Links ] https://doi.org/10.1016/j.lfs.2020.117839 [E-pub ahead of print].
5. Berlin D A, Gulick R M, Martinez F J. Severe Covid-19 . N Engl J Med. 2020 May 15. [ Links ] DOI: 10.1056/NEJMcp2009575 [E-pub ahead of print].
6. Bhatraju P K, Ghassemieh B J, Nichols M, Kim R, Jerome K R, Nalla A K, et al. Covid-19 in critically ill patiente in the Seattle region - case series. N Engl J Med. 2020; 382(21): 2012-22. [ Links ]
7. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T., et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020; 5(7): 1-8. [ Links ]
8. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497-506. [ Links ] https://doi.org/10.1016/S0140-6736(20)30183-5
9. Murray P R, Rosenthal K S, Pfaller M A Microbiología médica. 8va ed. Barcelona: Elsevier; 2017.p.506-511.
10. Fehr A.R, Perlman S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol Biol. 2015; 1282:1-23. [ Links ] https://doi.org/10.1007/978-1-4939-2438-7_1
11. Vellingiri B., Jayaramayya K., Iyer M., Narayanasamy A., Govindasamy V., Giridharan B., et al. COVID-19: A promising cure for the global panic. Sci Total Environ. 2020; 725:1-18. [ Links ]
12. D’Amico F., Baumgart D.C., Danese S., Peyrin Biroulet L. Diarrhea During COVID-19 Infection: Pathogenesis, Epidemiology, Prevention, and Management. Clin Gastroenterol Hepatol.2020; 18(8): 1663-72. https://doi.org/10.1016/j.cgh.2020.04.001
13. Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020; 181(2): 271-80. [ Links ]
14. Coutard B, Valle C, de Lamballerie X, Canard B, Seidah N G, Decroly E. The spike glucoprotein of the new coronavirus 2019-CoV containsa furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020; 176: 1-5. [ Links ] https://doi.org/10.1016/j.antiviral.2020.104742
15. Kim Y., Jedrzejczak R, Maltseva N I , Wilamowski M, Endres M, Godzik A,et al. Crystal structure of Nsp15 endoribonucleasa NendoU from SARS-CoV-2. Protein Sci. 2020; 29:1596-1605. [ Links ] https://doi.org/10.1002/pro.3873
16. Oliva Marín J.E. SARS-CoV-2 origen, estructura, replicación y patogénesis. Alerta. 2020; 3(2):79-86. [ Links ] https://doi.org/10.5377/alerta.v3i2.9619
17. Frieman M., Baric R. Mechanisms of Severe Acute Respiratory Syndrome Pathogenesis and Innate Immunomodulation. Microbiol Mol Biol Rev. 2008; 72(4): 672-85. [ Links ] https://doi.org/10.1128/MMBR.00015-08
18. Giwa A, Desai A., Duca A. Novel 2019 Coronavirus SARS-CoV-2 (COVID-19): An Overview for Emergency Clinicians. Emerg Med Pract. 2020; 22(5): 1-24. [ Links ]
19. Blanco-Melo D., Nilsson-Payant B.E, Liu W.C, Uhl S., Hoagland D., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19, Cell. 2020; 181 (5): 1036-45. https://doi.org/10.1016/j.cell.2020.04.026
20. Janeway C.A., Jr., Medzhitov R. Innate Immune Recognition. Annu Rev Immunol. 2002; 20 : 197-216. https://doi.org/10.1146/annurev.immunol.20.083001.084359
21. Lazear H.M., Schoggins J.W., Diamond M S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity. 2019; 50(4): 907-923. [ Links ]
https://doi.org/10.1016/j.immuni.2019.03.025
22. Chu H., Chan J. F-W., Wang Y., Yuen T. T-T., Chai Y., Hou Y., et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis. 2020 April 9. [ Links ] DOI: 10.1093/cid/ciaa410. ciaa4. [E-pub ahead of print]. https://doi.org/10.1093/cid/ciaa410
23. Li H., Liu L., Zhang D., Xu J., Dai H., Tang N., Su X., et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020; 395: 1517-20. [ Links ] https://doi.org/10.1016/S0140-6736(20)30920-X
24. Xu Z., Shi L., Wang Y., et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 2020; 8: 420-22 . [ Links ] https://doi.org/10.1016/S2213-2600(20)30076-X
25. Grossman S.C, Porth C.M. Fisiopatología. Alteraciones en la salud. Conceptos básicos. 9na ed. España : Wolters Kluver; 2014. p. 988-92.
26. Iba T., Levy J.H., Levi M., Connors J.M., Thachil J. Coagulopathy of Coronavirus Disease 2019. Crit Care Med. 2020 May 26; [ Links ] Doi: 10.1097 / CCM.0000000000004458 [E-pub ahead of print].
27. López-Salvio Y.M., Herrera-Rodríguez L.J., Guzmán-Silahua S., Nava-Zavala A.H., Rubio-Jurado B. Dímero D: papel en patología trombótica. El Residente. 2018; 13(1): 12-22. [ Links ]
28. Klok F.A., Kruip M.J.H.A., van der Meer N.J.M., Arbous M.S., Gommers D.A.M.P.J., Kant K.M.,et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thrombosis Research. 2020; 191: 145-47. [ Links ] https://doi.org/10.1016/j.thromres.2020.04.041
29. Delabranche X., Helms J., Meziani F. Immunohaemostasis: a new view on haemostasis during sepsis. Ann Intensive Care. 2017; 7(1): 117. [ Links ]
https://doi.org/10.1186/s13613-017-0339-5
30. Giannis D., Ziogas I.A., Gianni P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS CoV-1, MERS-CoV and lessons from the past. J Clin Virol. 2020 June. [ Links ] https://doi.org/10.1016/j.jcv.2020.104362 [E-pub ahead of print]. https://doi.org/10.1016/j.jcv.2020.104362
31. VCampbell C.M., Kahwash R. Will Complement Inhibition Be the New Target in Treating COVID-19-Related Systemic Thrombosis?. Circulation. 2020; 141(22): 1739-41. [ Links ]
32. Danzi G.B., Loffi M., Galeazzi G., Gherbesi E. Acute pulmonary embolism and COVID-19 pneumonia: a random association?. Eur Heart J. 2020; 41(19): 1858. [ Links ] https://doi.org/10.1093/eurheartj/ehaa254
33. Thachil J., Tang N., Gando S., Falanga A., Cattaneo M., Levi M., Clark C., et al. ISTH Interim Guidance on Recognition and Management of Coagulopathy in COVID-19. J Thromb Haemost. 2020; 18(5): 1023-26. [ Links ]
34. Thachil J. The versatile heparin in COVID-19. J Thromb Haemost. 2020; 18:1020-22. [ Links ]
35. Cheng Y., Luo R., Wang K., Zhang M., Wang Z., Dong L., et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020; 97: 829-38. [ Links ]
36. Fanelli V., Fiorentino M., Cantaluppi V., Gesualdo L., Stallone G., Ronco C., et al. Acute kidney injury in SARS-CoV-2 infected patients. Crit Care. 2020; 24: 155. [ Links ]
37. Wilson J.G., Calfee C.S. ARDS Subphenotypes: Understanding a Heterogeneous Syndrome. Crit Care. 2020; 24: 102. [ Links ]
38. Xiu-wu P., Da X., Hao Z., Wang Z., Lin hui W., Xin gang C.. Identifcation of a potential mechanism of acute kidney injury during the COVID-19 outbreak: a study based on single-cell transcriptome analysis. Intensive Care Med. 2020 March 31; [ Links ] DOI: 10.1007/s00134-020-06026-1 [E-pub ahead of print].
39. Hirsch J.S., Ng J.H., Ross D.W., Sharma P., Shah H.H., Barnett R.L., et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020; 98(1): 509-12. [ Links ]
40. Larsen C.P., Bourne T.D., Wilson J.D., Saqqa O., Sharshir M.A. Collapsing Glomerulopathy in a Patient With Coronavirus Disease 2019 (COVID-19). Kidney Int Rep. 2020; 5: 935-39. [ Links ]
41. Chan J. F-W., Yuan S., Kok K-H., Kai-Wang K., Chu H., et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020; 395 (10223): 514-523. [ Links ]
42. Gu J., Han B., Wang J. COVID-19: Gastrointestinal Manifestations and Potential Fecal-Oral Transmission. Gastroenterology. 2020; 158(6): 1518-19. [ Links ]
43. Li Z., Liu T., Yang N., Han D., Mi X., Li Y., et al. Neurological manifestations of patients with COVID-19: potential routes of SARS-CoV-2 neuroinvasion from the periphery to the brain. Front Med. 2020 May 4. [ Links ] https://doi.org/10.1007/s11684-020-0786-5 [E-pub ahead of print].
44. Netland J., Meyerholz D.K., Moore S., Cassell M., Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol. 2008; 82(15): 7264-75. [ Links ]
45. Iroegbu J.D., Ifenatuoha C.W., Ijomone O.M. Potential neurological impact of coronaviruses: implications for the novel SARS-CoV-2. Neurol Sci. 2020 May 18. [ Links ] https://doi.org/10.1007/s10072-020-04469-4 [E-pub ahead of print].
46. Zubair A.S., McAlpine L.S., Gardin T., et. al. Neuropathogenesis and Neurologic Manifestations of the Coronaviruses in the Age of Coronavirus Disease 2019. JAMA Neurol. 2020; 77 (8): 1018-27. [ Links ]
47. Baig A.M., Khaleeq A., Ali U., Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020; 11(7):995-98. [ Links ]
48. Sankowski R., Mader S., Valdés-Ferrer S.I. Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front Cell Neurosci. 2015; 9(28): 28. [ Links ]
49. Toscano G., Palmerini F., Ravaglia S., et al. Síndrome de Guillain-Barré asociado con SARS-CoV-2. N Engl J Med. 2020; 382 (26): 2574-76. [ Links ]
50. Galván Casas C., Català A., Carretero Hernández G., Rodríguez‐Jiménez P., Fernández Nieto D., Rodríguez‐Villa Lario A., et al. Classification of the cutaneous manifestations of COVID‐19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br J Dermatol. 2020; 183(1): 71-7. [ Links ]
51. Marzano A.V., Cassano N., Genovese G., Moltrasio C., Vena G.A. Cutaneous manifestations in patients with COVID‐19: A preliminary review of an emerging issue. Br J Dermatol. 2020 June 1; [ Links ] DOI: 10.1111/BJD.19264. . [E-pub ahead of print].
52. Sachdeva M., Gianotti R., Shah M., Lucia B., Tosi D., Veraldi S., et al. Cutaneous manifestations of COVID-19: Report of three cases and a review of literature. J Dermatol Sci. 2020; 98(2): 75-81. [ Links ]
53. Gianotti R., Zerbi P., Dodiuk-Gad R.P. Clinical and Histopathological study of skin dermatoses in patients affected by COVID-19 infection in the Northern part of Italy. J Dermatol Sci. 2020; 98(2): 141-3. [ Links ] https://doi.org/10.1016/j.jdermsci.2020.04.007
54. Babapoor-Farrokhran S., Gill D., Walker J., et al. Myocardial injury and COVID-19: Possible mechanisms. Life Sci. 2020 April 28; [ Links ] DOI: https://doi.org/10.1016/j.lfs.2020.117723 [E-pub ahead of print]. https://doi.org/10.1016/j.lfs.2020.117723
55. Burrel L.M., Risvanis J., Kubota E., Dean R.G., MacDonald P.S., Lu S., et al. Myocardial infartion increases ACE2 expression in rat and humans. Eur Heart J. 2020; 26(4): 369-75. [ Links ] https://doi.org/10.1093/eurheartj/ehi114
56. Dong M., Zhang J., Ma X., Tan J., Chen L., Xin Y., Zhuang L. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. Biomed Pharmacother. 2020 August 24; DOI: https://doi.org/10.1016/j.biopha.2020.110678 [E-pub ahead of print].
57. Boukhris M., Hillani A., Azzalini L. Cardiovascular Implications of the COVID-19 Pandemic: A global Perspective. Can J Cardiol. 2020; 36(7): 1068-80. [ Links ] https://doi.org/10.1016/j.cjca.2020.05.018
58. Graham Atkinson J. Problems with the analysis in “Treatment with Hydroxychloroquine, Azithromycin, and Combination in Patients Hospotalized with COVID-19”. Int J Infect Dis. 2020; 99:37.
59. Zou F., Qian Z., Wang Y., Zhao Y., Bai J. CJC Open. 2020 June 23; [ Links ] DOI: https://doi.org/10.1016/ j.cjco.2020.06.010. [E-pub ahead of print].
60. Tavazzi G., Pellegrini C., Maurelli M., Belliato M., Sciutti F., Bottazzi A., et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur J Heart Fail. 2020; 22(5): 911-15. [ Links ] https://doi.org/10.1002/ejhf.1828
61. Babapoor-Farrokhran S., Tarighati R., Gill D., Babapoor S., Amanullah A. Arrhythmia in COVID-19. Sn Compr Clin Med. 2020 August 14; [ Links ] DOI: 10.1002/ejhf.1828 [E-pub ahead of print]. https://doi.org/10.1002/ejhf.1828
62. Villamañán E., Armada E., Ruano M. Drug-induced QT interval prolongation: Do we know the risks?. Clinic Med. 2020; 144(6): 269-74. [ Links ]
63. Sánchez-Recalde A., Solano-López J.,Miguelena-Hycka J., Martín-Pinacho J.J., Sanmartín M., Zamorano J.L. COVID-19 and cardiogenic shock. Different cardiovascular presentations with high mortality. Rev Esp Cadiol. 2020; 73(8): 669-72. [ Links ] https://doi.org/10.1016/j.recesp.2020.04.018.

