










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkGaceta Médica Boliviana
On-line version ISSN 1012-2966
Gac Med Bol vol.38 no.1 Cochabamba June 2015
Caso Clínico
Encefalomiopatía mitocondrial, acidosis láctica y episodios de accidente cerebrovascular, síndrome de MELAS. Reporte de un caso clínico
Mitochondrial encephalomyopathy lactic acidosis and stroke like episodes MELAS syndrome. Report of a clinical case
Carlos Eduardo Padín1,a,b, Esteban Raúl Zirulnik1,b, Cecilia Rita Abraham1,c, Enrique Gonzalo Rojas Salazar3,d
1Fundación Escuela de Medicina Nuclear “FUESMEN" Mendoza, Argentina.2Servicio de Diagnóstico por Imágenes, Hospital del Carmen-OSEP.Mendoza Argentina.3Facultad de Medicina, Universidad Mayor de San Simón, Cochabamba, Bolivia.Jefe del servicio de tomografía “FUESMEN"; bMédico Radiólogo; cResidente de Radiología;d Médico Cirujano.*Correspodencia a: Enrique Gonzalo Rojas Salazar.Correo electrónico: enroque.rojas@gmail.com
Recibido el 02 de marzo de 2015.
Aceptado el 06 de abril de 2015.
Resumen
Las enfermedades mitocondriales producen una serie de desórdenes neurológicos que se heredan por parte materna, el síndrome de MELAS es considerado un raro desorden multisistémico neurodegenerativo de muy mal pronóstico, posee una incidencia de 16,3/100 000 casos, este síndrome se manifiesta antes de los 40 años, caracterizado por cuadros convulsivos, alteración del estado de conciencia, acidosis láctica, y accidentes cerebrovasculares, estas manifestaciones suelen ser evidentes en los estadíos avanzados, lo que dificulta su diagnóstico; siendo necesario un equipo multidisciplinario, donde los estudios de laboratorio y las técnicas imagenológicas juegan un papel fundamental. Les presentamos el caso de un paciente masculino de 29 años que acudió a emergencia del Hospital Central de Mendoza-Argentina con antecedentes de madre y hermana fallecidas. Tanto la tomografía como la resonancia magnética evidenciaron zonas infartadas de localización temporo-parieto-occipital, acompañado de calcificaciones en los núcleos basales, llegando al diagnóstico de síndrome de MELAS, para el cual no existe un tratamiento definitivo sólo paliativo.
Palabras claves: MELAS, enfermedades mitocondriales, acidosis láctica, resonancia magnética, Tomografía computarizada.
Abstract
Mitochondrial diseases are neurological disorders that are inherited maternally, the MELAS syndrome is considered a rare multisystem neurodegenerative disorder with a poor prognosis, has an incidence of 16,3 / 100 000 cases, this syndrome is manifested before age 40 years, characterized by convulsive, altered state of consciousness, lactic acidosis, and stroke, these manifestations are usually evident in advanced stages, making it difficult to diagnosis; being necessary equipment multi-disciplinary where laboratory studies and imaging techniques play a fundamental role. We present the case of a male patient of 29 years, who attended in Emergency of Hospital Central Mendoza-Argentina with a history of mother and sister dead. Both Computed tomography and Magnetic resonance showed infarcted areas with localization temporo-parieto-occipital, accompanied by calcifications in the basal nuclei, reaching the diagnosis of MELAS. for this syndrome there is not definitive treatment, only palliative.
Keywords: MELAS, mitochondrial diseases, lactic acidosis, magnetic resonance, computed tomography.
El síndrome de Encefalomiopatía Mitocondrial, Acidosis Láctica y Accidentes Cerebrovasculares (MELAS) es considerado un raro desorden multisistémico neurodegenerativo de muy mal pronóstico, de herencia materna y es causado por mutaciones en el DNA mitocondrial1,2,3, tiene una incidencia de 16,3/100 000 casos 4,5, fue descrito inicialmente por Shapira y col. en el año 1 975, posteriormente Pavlakis en 1 984 fue el primero en utilizar el acrónimo MELAS6.
La mitocondria es esencial para el metabolismo energético celular, su principal función es la obtención de energía por medio de la fosforilación oxidativa para producir ATP7,8, la etiología de este síndrome se debe a diferentes mutaciones puntuales del ADNmt siendo la más frecuente es la sustitución de A por G en el gen ARNt leu(UUR) en el nucleótido 3 243, presente en el 80-85% de los casos9-12. Las mitocondrias son heredadas del lado materno, puesto que en el gameto masculino la mayor parte de éstas se encuentran localizadas en la cola del espermatozoide, por ende no participan en la fertilización. Las madres afectadas con alguna mitocondrio-patía heredarán el carácter a su descendencia, no sucediendo esto con los padres afectados1,13.
Los pacientes con MELAS desarrollan disfunción cerebral, dilatación de los ventrículos, atrofia cortical, calcificación de los ganglios de la base e infartos cerebrales1 2 1 2 3. Estos infartos tienen la característica de no ser vasculares, son causados por la disfunción en la fosforilación oxidativa dentro del parénqui-ma cerebral6 dichas características son detectadas mediante estudios complementarios como la Tomografía Computarizada (TC), y Resonancia Magnética (RM) cerebral2,6.
Los reportes de esta patología describen múltiples manifestaciones clínicas, todos ellos coinciden con tres características clínicas invariables dentro de este síndrome: 1) episodios de accidente cerebrovascular antes de los 40 años, 2) la encefalopatía caracterizada por presentar crisis convulsivas, demencia, o ambas, 3) acidosis láctica, o por medio de una biopsia muscular la presencia de fibras rojo rasgadas o ambas. Esta es la triada que distingue clínicamente al síndrome de MELAS de las demás enfermedades mitocondriales14,15.
Este síndrome puede manifestarse clínicamente por retardo en el desarrollo, baja estatura, dificultad en el aprendizaje, déficit de atención, migraña, deterioro cognitivo lento y progresivo hasta llegar a la demencia1, del mismo modo pueden presentar hipotonía, debilidad muscular, intolerancia al ejercicio, cansancio fácil, y cardiomiopatía hipertrófica. Puede existir afectación a los sistemas sensoriales presentando hemiplejía, oftalmoplejía, atrofia óptica, retinitis pigmentaria, y sordera neurosensorial, esta patología suele asociarse con defectos en la secreción de insulina y oliguria relacionados con el síndrome nefrítico6-16.
Estas manifestaciones no son todas exclusivas de las enfermedades mitocondriales, sin embargo una combinación de tres o más de estos síntomas constituye una fuerte sospecha de una patología mitocondrial fundamentalmente cuando afectan a varios órganos15, 17. Los órganos más afectados son aquellos que requieren mayor demanda energética y por tanto mayor número de mitocondrias, como el músculo esquelético y cardíaco, el riñón, el páncreas, el hígado, la retina y el sistema nervioso central6,8,18-20, una vez presentados los síntomas iniciales el síndrome presenta un curso evolutivo rápido5,10.
Ante una sospecha de este síndrome es fundamental realizar una TC y RM que junto con la clínica, y los antecedentes familiares fundamenten su diagnóstico, para posteriormente realizar un tratamiento que pueda prolongar la supervivencia y mejorar la calidad de vida de estos pacientes21, ya que actualmente no existe un tratamiento curativo específico aprobado para esta patología, se han empleado diversas alternativas para detener el progreso y el deterioro de los pacientes, cuyas terapias pretendían incrementar la producción de ATP utilizando coenzima Q10, corticoesteroides, biotina, ácido fólico, L-carnitina, tiamina, rivoflavina, vitamina C, entre otros, dichas alternativas terapéuticas presentaron poco éxito21-23.
Presentación del caso
Paciente masculino de 29 años de edad ingresa al servicio de emergencias del Hospital Central de Mendoza-Argentina en septiembre de 2 014 presentando cefalea, convulsiones,y alteración del estado de conciencia. Presenta antecedentes de convulsiones de larga data y antecedentes heredofamilia-res de importancia; con madre y hermana fallecida antes de los cuarenta años, según reportes ambas presentaron cuadros convulsivos con alteraciones motoras y mentales antes de su muerte.
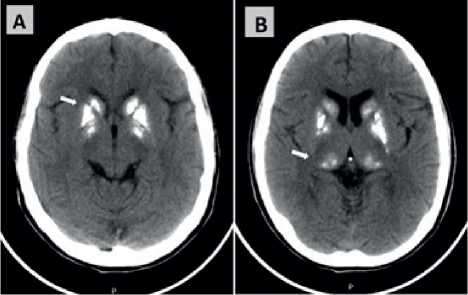
En el servicio de emergencias se le solicitó una TC de cerebro que evidenció a nivel de los núcleos de la base imágenes de densidad cálcica, a nivel de la cabeza del núcleo caudado, núcleo lenticular y del tálamo óptico en forma bilateral y simétrica (Figura 1 A y B). Los exámenes de laboratorio se encontraban dentro de los parámetros normales. Debido al cuadro convulsivo, los antecedentes familiares y las calcificaciones a nivel de los núcleos de la base, se sospechó el diagnóstico de Síndrome de Fahr, que es una rara patología con las características clínicas e imagenológicas mencionadas.
En noviembre del mismo año el paciente ingresó nuevamente al servicio de emergencias presentando un cuadro de crisis convulsiva, alteración del estado de conciencia, lenguaje incoherente, familiares refieren la disminución del peso corporal y de la ingesta de líquidos. Al examen neurológico el paciente se encontró vigil, pupilas isocóricas fotorreactivas; sin signos de foco motor ni meningismo, presentó trastornos conductuales y excitación psicomotriz, incapacidad de responder a órdenes simples, los signos vitales se encontraron dentro de los parámetros normales. Por las características mencionadas se decide su internación.
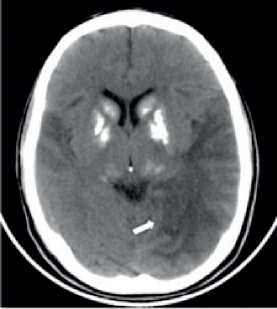
Figura 2. Tomografía Computarizada sin contraste que evidencia: Hipodensidad cortico-subcortical temporo-parieto-occipital del lado izquierdo y en menor medida a nivel temporo-parietal del lado derecho.
Se solicitó exámenes de laboratorio y una nueva TC, el resultado laboratorial más relevante fue el ácido láctico elevado en sangre venosa 5 mmol/L (Valor normal 0,5 a 2,2 mmol/L), leucocitos de 14 200mm3 (Valor normal 5 000 a 10 000mm3), tanto la glucemia, calcio, potasio, y sodio se encontraron dentro los valores normales.
La nueva tomografía detecta hipodensidad cortico-sub-cortical a nivel temporo-parieto-occipital del lado izquierdo y en menor medida a nivel temporo-parietal del lado derecho. Con la sospecha de un ictus agudo se le realizó una RM en la que se evidenció: Lesiones cortico-subcorticales a nivel tem-poro-parieto-occipital del lado izquierdo y temporo-parietal del lado derecho, con señal hipointensa en T1 e hiperintensa
en T2, FLAIR y difusión, con edema de las circunvoluciones, y compresión de los dos surcos corticales. La lesión del lado izquierdo presentó en nuestro paciente un ligero “efecto de masa” sobre el sistema ventricular homolateral produciendo colapso parcial del asta occipital del ventrículo lateral (Figura 3). Dichas imágenes son compatibles con lesiones pseudoic-tales corticales, también se evidenciaron las calcificaciones en los ganglios de la base ya descritas en la TC.
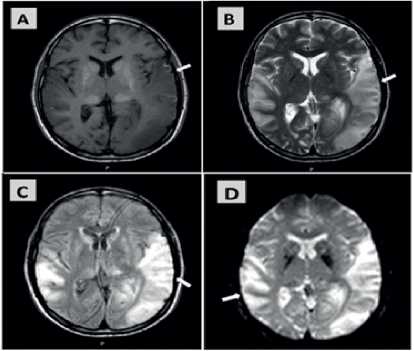
Figura 3. Cortes axiales de RM en secuencias T1 (Imagen A), T2 (imagen B), FLAIR (Imagen C) y Difusión (Imagen D) que evidencian lesiones cortico-subcorticales a nivel temporo-parieto-occipital del lado izquierdo y temporo-parietal del lado derecho, con señal hipointensa en T1, e hiperintensa en T2, FLAIR y difusión, con edema de las circunvoluciones y compresión de los surcos corticales.
Ante los antecedentes heredofamiliares, la evolución neu-rológica, la edad del paciente, la presencia de acidosis láctica, las calcificaciones de los ganglios de la base, y el accidente ce-rebrovascular evidenciado por la RM, se confirma un síndrome de MELAS. Lamentablemente no existe un tratamiento definitivo ni cura para este síndrome, y una vez iniciada la lesión cerebrovascular, el cuadro sigue un curso rápido y de mal pronóstico, por lo que el paciente permanece internado con medidas higiénico dietéticas permanentes brindándole de ese modo una mejor calidad de vida.
Discusión
El pronóstico del síndrome MELAS es malo, y progresa con múltiples déficits neurológicos y finalmente la muerte, puede ser de origen esporádico o familiar; que en dicho caso es de transmisión materna3,24 . La presentación clínica del síndrome de MELAS es muy variable, y se asocia a accidentes cerebrovasculares antes de los 40 años, aunque se han reportados casos de pacientes con edad avanzada6,24-26.
Los niveles elevados de ácido láctico pueden encontrarse dentro de los valores normales o levemente aumentados hasta las fases avanzadas, dificultando de este modo un diagnóstico precoz6. Acorde a la clínica de nuestro paciente, los antecedentes familiares de una madre y hermana fallecidas habiendo presentado cuadros convulsivos, nos orientaron a una patología de origen genético, las primeras imágenes ya descritas mostraron calcificaciones en los ganglios basales, pero no así lesiones infartadas, los laboratorios iniciales no mostraron alteraciones en el ácido láctico, por lo que no teníamos un diagnóstico preciso.
En cuanto a los estudios imagenológicos los hallazgos en la TC son inespecíficos, en los cuales se pueden evidenciar zonas infartadas de localización parieto-occipital afectando a los núcleos de la base que pueden presentar calcificaciones o in-fartos5,6. En la RM las imágenes no son patognomónicas, pero cumplen un patrón característico de este síndrome, sobre todo por la presencia de infartos corticales temporo-occipitales sin respetar los territorios vasculares y la calcificación bilateral y simétrica de los ganglios basales, junto con la hiperintensidad en secuencias ponderadas en T1, T2 y FLAIR en forma simétrica de ambos pulvinares5, aunque se han reportado casos en los que la RM se encuentra normal6.
En relación a nuestro paciente fue en la crisis convulsiva, la excitación psicomotriz y los trastornos de conciencia, cuando la última TC evidenció zonas infartadas a nivel temporo-pa-rieto-occipital del lado izquierdo y en menor medida a nivel temporo-parietal del lado derecho, la RM demostró también hiperintensidad en T2, y FLAIR, que acorde a la literatura corresponden a imágenes propias de esta patología, que junto con la clínica y la presencia de acidosis láctica, se llegó al diagnóstico definitivo de síndrome de MELAS.
Actualmente no existe un tratamiento curativo específico para este síndrome, ya que se han utilizado diversos tratamientos y drogas con la esperanza de detener el progreso y deterioro de los pacientes, junto con terapias para lograr un incremento en la producción de ATP como Coenzima Q10, corticoesteroides, ácido fólico, tiamina, vitamina C, etc., con poco éxito7,22,23.
Podemos concluir que el síndrome de MELAS debe ser considerado como diagnóstico diferencial en los pacientes jóvenes que presentaron infartos cerebrales con predominio en la zona cortical y localización temporo-parieto-occipital que no se adapten a un solo territorio vascular, y que para conseguir un diagnóstico de certeza es necesario un enfoque multidisciplinario, siendo las imágenes una herramienta fundamental, acompañadas de los controles y la evolución del paciente.
Referencias bibliográficas
1. Espinoza-López DA, Vargas-Cañas ES, Díaz-Alba A, Morales-Briceño H, Ramírez-Jiménez C, Fernández-Valverde F, et al. Encefalopatía mito-condrial, acidosis láctica y episodios stroke like (MELAS). Arch Neurocien (Mex). 2012; 17(2): 138-41
2. García L, Guglielmo R, Illa L. Síndrome de ME-LAS: TC y RM como herramienta diagnóstica no invasiva. Revista argentina de radiología. 2010; 74(4): 379-83.
3. Cano A, Romero AI, Bravo F, Vida JM, Espejo S. Síndrome MELAS: hallazgos neurorradiológicos. Radiología. 2002; 44(2): 69-74.
4. Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, et al. Prevalence of mitochondrial DNA disease in adults. Annals of neurology. 2008; 63(1): 35-9.
5. McFarland R, Taylor RW, Turnbull DM. The neurology of mitochondrial DNA disease. The Lancet Neurology. 2002; 1(6): 343-51.
6. Guevara-Campos J, Gonzalez-Guevara L, Urbáez-Cano J, Parada Y. Encefalopatía infantil asociada con la mutación A3243G MELAS. Investigación Clínica. 2007; 48(2).
7. Schapira AH. Mitochondrial disease. The Lan-cet. 2006; 368(9529): 70-82.
8. Ruiz-Siebald P-P, Canales P. Enfermedades mi-tocondriales: diagnóstico diferencial de enfermedad cerebrovascular en adulto joven a propósito de un caso. Revista chilena de neuro-psiquiatría. 2013; 51(1): 25-31.
9. Seijo ÁG, Orjales MC, Benavent JP. MELAS: claves del diagnóstico y tratamiento en la Unidad de Cuidados Intensivos. Medicina intensiva. 2008; 32(3): 147-50.
10. Coelho-Miranda L, Playan A, Artuch R, Vi-laseca M, Colomer J, Briones P. Encefalopatía mitocondrial, acidosis láctica y accidentes cere-
brovasculares (MELAS) en edad pediátrica con la mutación A3243G en el gen ARNtLeu (UUR) del ADN mitocondrial. Rev Neurol. 2000; 31: 804-11.
11. Tapia-Pérez J, Rodríguez-Leyva I, Oros-Ova-lles C. Síndrome de sobreposición MELAS/ME-RRF: Informe de un caso y revisión de la literatura. Rev Mex Neuroci. 2003; 4(5): 360-5.
12. Marín MVP, Carrisoza J, Pérez PF, Cornejo JW, Ruiz A, Bedoya G. Genética de la citopatía mitocondrial MELAS (Encefalomiopatía Mito-condrial, Acidosis Láctica y Apoplejía).
13. Pakendorf B, Stoneking M. Mitochondrial DNA and human evolution. Annu Rev Genomics Hum Genet. 2005; 6: 165-83.
14. Muñoz-Nevárez LA, Martín-Nares E, Ontive-ros-Mercado H, Alvarado-Verduzco H, Valerdi-Contreras L, Ramírez-Guzmán MG. MELAS: una serie de casos del Hospital Civil de Guadalajara «Fray Antonio Alcalde». Revista de Endocrinología y Nutrición. 2013; 21(3): 138-47.
15. Hirano M, Ricci E, Koenigsberger MR, Defen-dini R, Pavlakis SG, DeVivo DC, et al. MELAS: an original case and clinical criteria for diagnosis. Neuromuscular Disorders. 1992; 2(2): 125-35
16. Abe K, Yoshimura H, Tanaka H, Fujita N, Hi-kita T, Sakoda S. Comparison of conventional and diffusion-weighted MRI and proton MR spectros-copy in patients with mitochondrial encephalom-yopathy, lactic acidosis, and stroke-like events. Neuroradiology. 2004; 46(2): 113-7.
17. Fernández-Valverde F, Tena-Suck ML, Vargas-Cañas S, Salinas-Lara C, García-Márquezd A, Co-llado-Ortize MÁ. Mujer de 17 años de edad con cefalea, ictus y crisis epilépticas. Gac Méd Méx. 2009; 145(5).
18. Cortés A, Alcázar M, Navas P, Rodríguez-Her-nández M, Asencio C. Diagnóstico genético de paciente con enfermedad mitocondrial. Biosaia: Revista de los másteres de Biotecnología Sanitaria y Biotecnología Ambiental, Industrial y Alimentaria. 2014; 1(3).
19. González TR, Jarque MV. Las enfermedades mitocondriales: un reto para las Ciencias Médicas. Medisan. 2004; 8(1): 43-50.
20. Haas R, Parikh S, Falk M. Enfermedad mito-condrial: abordaje práctico para los médicos de atención primaria. Pediatrics. 2007; 64(6): 321-8.
21. Espinoza-López DA, Vargas-Cañas ES, Díaz-Alba A, Morales-Briceño H, Ramírez-Jiménez C, Fernández-Valverde F, et al. Encefalopatía mito-condrial, acidosis láctica y episodios stroke like (MELAS). Arch Neurocien (Mex). 2012; 17(2): 138-41.
22. Singh B, Low P, Yeo J. MELAS: a case report. ANNALS-ACADEMY OF MEDICINE SINGA-PORE. 2004; 33: 69-71.
23. Berbel-Garcia A, Barbera-Farre JR, Etessam JP, Salio AM, Cabello A, Gutierrez-Rivas E, et al. Coenzyme Q 10 improves lactic acidosis, stroke-like episodes, and epilepsy in a patient with ME-LAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes). Clinical neuropharmacology. 2004; 27(4): 187-91.
24. Kimata KG, Gordan L, Ajax ET, Davis PH, Grabowski T. A case of late-onset MELAS. Archives of neurology. 1998; 55(5): 722-5.
25. Hirano M, Pavlakis SG. Topical review: mito-chondrial myopathy, encephalopathy, lactic aci-dosis, and strokelike episodes (MELAS): current concepts. Journal of child neurology. 1994; 9(1): 4-13.
26. Sharfstein SR, Gordon MF, Libman RB, Mal-kin ES. Adult-onset MELAS presenting as herpes encephalitis. Archives of neurology. 1999; 56(2): 241-3.

