










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkGaceta Médica Boliviana
Print version ISSN 1012-2966On-line version ISSN 2227-3662
Gac Med Bol vol.37 no.2 Cochabamba Dec. 2014
Caso Clínico
Tumor de Wilms unilateral asociado a aniridia: a propósito de un caso
Wilms tumor associated with unilateral aniridia: a purpose of a case
Mayra Victoria Rocha Choque1,2, Alejandro Méndez Pardo2, Claudia Terrazas Saavedra2, Antonio Jose Pardo Novak3
1Auxiliar del Departamento de Educación Médica y Planificación (DEMyP). 2Estudiante de Medicina, Facultad de Medicina de la Universidad Mayor de San Simón. Cochabamba, Bolivia. 3M.D. - Médico Ginecólogo-Obstetra. Jefe de Guardia, Hospital Materno Infantil German Urquidi. Docente Pre y Post-Grado Facultad de Medicina-Universidad Mayor de San Simón. Cochabamba, Bolivia.*Correspodencia a: Mayra Victoria Rocha Choque Correo electrónico: mayita_9_2@hotmail.com
Recibido el 17 de agosto de 2014.
Aceptado el 22 de septiembre de 2014.
Resumen
El Nefroblastoma es el tumor renal más frecuente en la infancia afectando a 1 de 10 000 niños en EEUU, la edad de los afectados más frecuente es de 2 a 5 años. Se presenta un paciente de 16 meses de edad, sexo masculino, que asistió a consulta externa pediátrica, por dolor en flanco izquierdo, además de presentar aniridia, solicitamos una tomografía de abdomen que evidencia un tumor en riñón izquierdo en polo inferior de 10 cm x 7 cm desplazando al órgano en sentido posterior y cefálico. La conducta fue que el paciente se sometiera a nefrectomía radical, el estudio histopatológico reveló tumor trifásico de histología favorable, tipificado en estadio I. El caso presentado cobra especial relevancia debido a que los casos publicados, en su mayoría tratan de tumores bilaterales los cuales más comúnmente están asociados a aniridia. El manejo de este tumor en general tiene buen pronóstico, teniendo algunas excepciones. El diagnóstico precoz debe basarse en el examen ocular exhaustivo al nacer, en caso de hallarse aniridia en el paciente, se debe realizar una ecografía abdominal para descartar la posibilidad un nefroblastoma renal.
Palabras claves: tumor de Wilms, nefroblastoma, aniridia.
Abstract
The Nephroblastoma is the most common renal tumor in childhood affecting 1 in 10,000 children in the United States, the age of those affected more often 2 to 5 years. A patient's 16-month-old male, who attended pediatric outpatient pain in left flank, besides presenting aniridia Presented requested a CT of the abdomen demonstrates a tumor in the left kidney tumor in the lower pole of 10cm x 7cm body displacing posteriorly and cephalad. The behavior was that the patient was submitted to radical nephrectomy,histopathological study revealed favorable histology tumor phase, typified Stage I. This case becomes relevant especially because reported cases, mostly dealing with bilateral tumors which more are commonly associated with aniridia. The management of this tumor generally has a good prognosis, with some exceptions. Early diagnosis should be based on thorough eye examination at birth, if found in aniridia patient should perform an abdominal ultrasound to rule out renal nephroblastoma.
Keywords: Wilms tumor, nephroblastoma, aniridia.
EL tumor de Wilms o nefroblastoma, es el tumor renal primario más frecuente de la primera infancia, afecta a 1 de cada 10 000 niños en EEUU y es el cuarto tumor maligno más frecuente en pediatría1.
La incidencia máxima está entre los 2 a 5 años de edad, el 90%, se presenta de forma unilateral, es frecuente asociar está neoplasia a malformaciones congénitas2. El uso actual de esquemas terapéuticos juiciosos ha llevado a una curación del tumor del 85%2. Es más frecuente en menores de raza negra y una razón de género 1:11,3.
El tumor se presenta con o sin antecedentes familiares, casi 97% se presentan de forma esporádica sin causa hereditaria, congénita o factores de riesgo3. Tanto en formas hereditarias y esporádicas se identifican mutaciones del gen WT1 localizado en el cromosoma 11p133. Es apreciable la teoría de los dos golpes para los genes supresores tumorales recesivos, la proteína WT1 está implicado en la diferenciación renal y gonadal, que regula el desarrollo del riñón, su deficiencia tiene un efecto oncógeno en la diferenciación genitourinaria, a pesar de ser un supresor tumoral su sobrexpresión actúa como un onco-gén causante de otras neoplasias en el adulto1-3. Existen grupos de pacientes que tienen tumor de Wilms conjuntamente con síndromes como: el síndrome WAGR (33%) y síndrome de Denys-Drash.
Clínicamente los pacientes pueden presentar una masa palpable asintomática en el abdomen de carácter uni o bilateral, cuando el tumor es muy grande puede llegar hasta la pelvis o ser vulnerable a traumatismos. Otros pacientes presentan una forma sintomática con hematuria, dolor, obstipación, reducción de peso e hipertensión2,3.
Macroscópicamente el tumor de Wilms se observa como una masa bien delimitada, grande y solitaria, color grisáceo pardo, blando y homogéneo con focos de necrosis, hemorragia y quistes ocasionales que crece en cualquier parte del riñón, en forma variable, y generalmente encapsulados. Microscópicamente se reconocen combinación de células del blastema, epitelio y estroma2. Solo un 5% de los tumores cursa con anaplasia con mutaciones de p53 y un pronóstico desfavorable con resistencia a la quimioterapia. La mayoría se presenta con características histológicas favorables en ausencia de anaplasia1,2.
Los restos nefrógenos pueden ser precursores del tumor de Wilms en un 25 a 40% de los tumores unilaterales y casi todos los bilaterales1.
En gran parte de los casos al momento del diagnóstico ya existen metástasis pulmonares. Los tumores sin características anaplasias se pueden curar4, lo que demuestra la importancia de verificar el tipo histológico. Se han descrito tumores secundarios o causados por la radioterapia1.
El tratamiento según la National Wilms Tumor Study Groupdebe ser una intervención quirúrgica seguida de quimioterapia, mientras que la International Society of Paediatric Oncology sugiere reducir el volumen tumoral con quimioterapia preoperatoria3. Si es unilateral se realiza una nefrourete-rotomia radical, en casos bilaterales se realiza quimioterapia seguida de una nefrectomía parcial con conservación de ne-fronas3, 5.
El objetivo de presentar este caso radica en las características clínicas que presenta el paciente con tumor de Wilms unilateral asociado a aniridia, el caso será un aporte para los datos epidemiológicos y futuros estudios sobre este tumor.
Presentación del caso
Paciente de consulta externa de pediatría, de sexo masculino con 16 meses de edad, raza mestiza. Los padres refieren que el niño manifiesta dolor en flanco izquierdo de 3 meses de evolución, progresivo y no asociado a trauma además de la presencia de una masa y aumento de volumen en la zona abdominal izquierda.
El paciente es producto de tercera gestación, embarazo sin complicaciones, de término, nacido por cesárea, APGAR 10/10, peso de 2 540 g, lactancia materna hasta la fecha, vacunas completas para la edad.
Los padres no refieren antecedente quirúrgico ni patológico, refieren que en meses previos presentaba episodios de fiebre y los últimos días anorexia e irritabilidad. No refieren antecedentes familiares de tumores malignos o malformaciones hereditarias.
Al examen físico, paciente consciente, orientado, eutrófico, afebril, hidratado e inquieto. Al examen ocular presenta aniridia bilateral con fotofobia. A la palpación superficial del abdomen se encuentra blando, depresible y no doloroso, en la palpación profunda en flanco izquierdo se palpa masa, no móvil, dolorosa, de consistencia dura, de aproximadamente 15 cm de diámetro, de contornos regulares con superficie lisa. Dicha masa se extiende de la línea axilar anterior hasta la línea axilar posterior, es positiva la puño percusión en flanco izquierdo. Signos vitales: FR: 32x min, FC: 110 x min, T°: 36.8 Peso: 10 kg, Talla: 80 cm, IMC: 15,63. El examen genitourinario evidencia estrechamiento del orificio prepucial y fimosis leve.
La ecografía abdominal muestra una imagen redondeada de aspecto sólido que aproximadamente mide 10 cm de diámetro sobre el polo inferior del riñón izquierdo. El hemograma presenta linfopenia, ligera eosinofilia y anemia ferropénica.
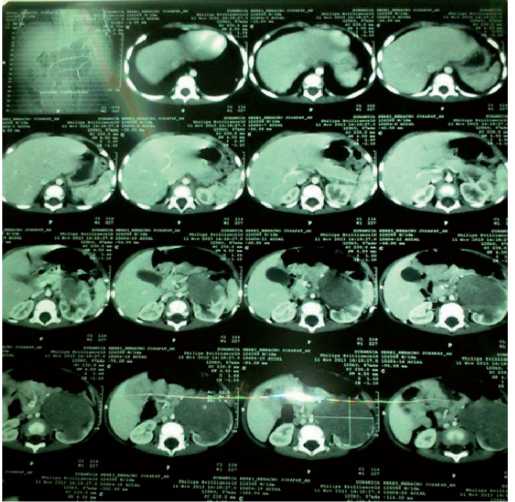
Se le realizó una Tomografía Axial Computarizada (TAC) abdominal el que evidencia masa hipodensa en el polo inferior del riñón izquierdo de un diámetro de 10 cm x 7 cm desplazando al riñón en sentido posterior y cefálico. El hígado, retroperitoneo y glándulas suprarrenales sin alteraciones. Diagnóstico altamente sugestivo de tumor de Wilms. TAC de tórax sin imágenes sugestivas de metástasis (Fig. 3).
El paciente fue sometido a una laparotomía exploratoria hallándose una masa compatible con tumor de Wilms, se procede a realizar nefrectomía radical izquierda tomando pieza para examen histopatológico que confirma el diagnóstico. Al finalizar la intervención quirúrgica se realiza reparación de la fimosis.
En el estudio histopatológico de la pieza quirúrgica esta pesa 197 gr y mide 10 x 7 x 6 cm. Macroscópicamente exhibe cápsula renal café clara de bordes bien circunscritos. Al corte se observa una masa tumoral de 8 cm de diámetro mayor, blanquecina grisácea, granular y blanda. En la periferia se observa corteza renal café clara y blanda. El hilio con segmento de uréter de 6 cm tubular y blando. La arteria y vena renal están seccionadas en el borde del hilio (Fig. 1-2). Microscópicamente los cortes histológicos muestran riñón con neoplasia embrionaria infiltrante, formada por nidos sólidos de células
Figura 1. Riñón izquierdo al corte se observa el tumor que ocupa aproximadamente 2/3 del polo inferior del parénquima renal.
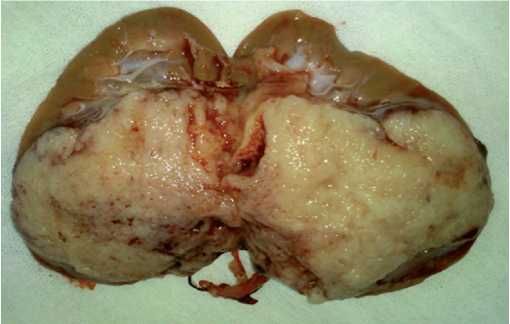
Figura 2. Riñón y tercio proximal del ureter izquierdo extraido tras la nefrectomia unilateral. Aspecto macroscópico del tumor.
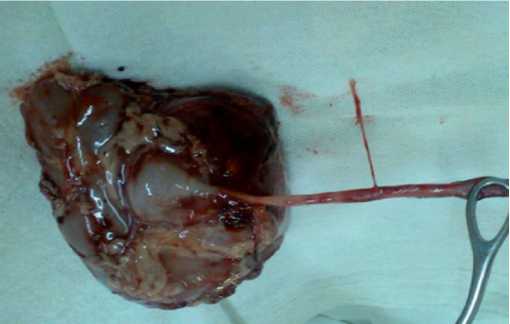
Figura 3. Estudio con medio de contraste en fase parenquimatosa. Se observa imagen hipodensa inmersa en el parénquima renal izquierdo.
pequeñas de bordes angulados, hipercromáticos, con distribución densa de la cromatina y mitosis escasas. No se identifica anaplasia. Componentes blastemal, estromal y epitelial de histología favorable.
Una vez identificada la masa, según la literatura revisada debe hacerse diagnóstico diferencial con Hidronefrosis, ne-fropatías quísticas, neuroblastomaintrarrenal, nefromameso-blastico y sarcoma renal. Al sospechar de una masa de origen renal, se debe hacer la diferenciación principalmente con el neuroblastoma, el cual pudo ser descartado por las características imagenológicas que caracterizan a cada uno de los tumores, dando más indicios por un tumor de Wilms por ser intrarrenal, márgenes bien definidos y bordes regulares como es usualmente.
Discusión
El síndrome de WAGR es un raro trastorno genético caracterizado por una deleción del cromosoma 11p13 y está clínicamente asociada con el tumor de Wilms, aniridia, anomalías genitourinarias y retraso mental (WAGR)6,7.
El tumor de Wilms es la neoplasia más frecuente en la infancia, pero su prevalencia en nuestro medio es incierta al no contarse con datos epidemiológicos, este tumor suele presentarse clínicamente con malformaciones congénitas del aparato genitourinario, otro punto importante es la presencia de aniridia esporádica o familiar, la literatura hace referencia que la presentación de aniridia en neonatos aumenta 257 veces la probabilidad de tener un nefroblastoma, y muchas veces resulta ser el motivo de consulta primario8. Se ha descrito la presencia de aniridia en pacientes con nefroblastomas bilaterales, lo cual contrasta con el caso que presentamos, que es un nefroblastoma de presentación unilateral con asociación de aniridia. La incidencia de la aniridia es de aproximadamente 1 en 50 000 en la población general8.
Figura 4. Estudio con medio de contraste en fase parenquimatosa. Se observa imagen hipodensa inmersa en el parénquima renal izquierdo.
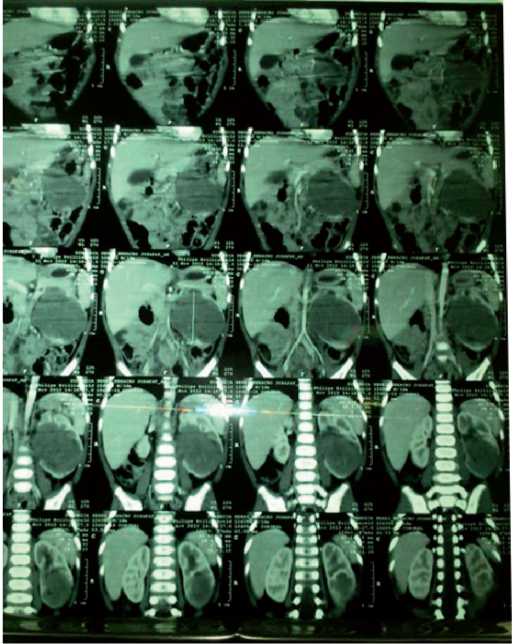
La alta asociación de aniridia esporádica con el tumor de Wilms exige un examen ocular exhaustivo al nacer junto con la ecografía renal, que se repetirá cada 3-6 meses. A la inversa de todos los casos de tumor de Wilms deben ser sometidos a un examen ocular para la detección de posibles anomalías oculares asociadas 6A9. Situación que no se hizo en el paciente por los recursos limitados de la familia.
Hablando sobre la tipificación del tumor el Grupo de Estudio sobre el Tumor de Wilms (NWTSG) lo clasifica en 4 estadios. El estadio I está limitado al riñón y la superficie capsular esta integra. El estadio II se extiende dentro del riñón pero pueden extirparse. El estadio III se limita a la cavidad abdominal. El estadio IV metástasis hematógenas, frecuentemente pulmón4,10. En el caso presentado por las características macro y microscópicas se tipifico el tumor en estadio 1, a diferencia de los casos revisado estos ya se presentaban a estadios avanzados.
En el caso se realizó una nefrectomía total izquierda sin linfadenectomia asociada lo cual respalda la literatura que indica que no existe evidencia clínica del beneficio sobre la realización de una linfadenectomia asociada. Existen diversas recomendaciones sobre el manejo de este tumor, como el NWTSG indica que se debe realizar una intervención quirúrgica seguida de quimioterapia como se realizó en este caso, otra opinión como la International Society of Paediatric Oncology indica que el manejo debería ser una quimioterapia preoperatoria para reducir el tamaño del tumor y facilitar la intervención quirúrgica, sin embargo esto dificulta la tipificación real el tumor3,9,10.
Independientemente de la terapéutica escogida el pronóstico es favorable, excepto los tumores bilaterales, anaplásicos y con estadios avanzados10,11.
García et-al sugieren que los niños con un peso combinado del riñón afectado y el tumor de menos de 550 gr tienen un buen pronóstico. Los niños con estadio I con una edad menor de 24 meses tuvieron un resultado excelente10.
Los pacientes con tumor de Wilms, con una historia clínica detallada y examen físico preciso llevan a un diagnóstico oportuno que aportan al paciente muchas oportunidades de
superar con éxito esta neoplasia.El método diagnóstico más útil en esta patología sigue siendo la tomografía y la ultraso-nografía, que también juegan un papel muy importante para la estatificación de esta neoplasia en el riñón.
El tratamiento más indicado para los pacientes con tumor de Wilms siempre es personalizado y encaminado en las guías de la National Wilms Tumor Study Group y la Society of Paediatric Oncology, sobre el uso de la quimioterapia y cirugía en todos los estadios. El uso juicioso de estos esquemas terapéuticos ha llevado a tasas de curación del 85% siempre que el diagnóstico definitivo brindado por el estudio histopatológico sea favorable, por lo que el pronóstico y evolución de estos pacientes son muy buenos en la mayoría de los casos.
Referencias
1. Robbins S, Cotran RS, Kumar V, Collins T. Patología estructural y funcional. 8va ed. Madrid: McGrau-Hill ELSEVIER, 2010.
2. Anirban Maitra. Enfermedades de la lactancia y la infancia. En Robbins S, Cotran RS, Kumar V, Collins T. Patología estructural y funcional. 8va ed. Madrid: McGrau-Hill ELSEVIER, 2010. P. 447-81.
3. Hackam D, Grikscheit T, Wang S, Newman D y Ford R. Cirugía Pediátrica. En: Schwartz, F. Charles Brunicardi. Principios de Cirugía. 9na edición. México. Mc Graw-Hill;2010 p.1409-1455.
4. Badrnath R, Konety MD, Richard D. Neoplasias del parénquima renal. En: Jack W, McANINCH, Lue T. Smith y Tanagho Urología general. 18va edición. Mexico. Mc Graw-Hill-Lange:2014 ;p. 342-46.
5. Hernández R. El tumor de Wilms. Un paradigma de heterogeneidad genética: a paradigm of geneticheterogeneity. Revhabanciencméd [revista en la Internet]. 2011 Jun [citado 2014 Jun 21] ; 10(2):Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext;pid=S1729-519X2011000200008;lng=es.
6. Fischbach BV, Trout K, Lewis J, Sika M, et al. WARG Syndrome: A Clinical Review of 54 Cases. Pediatrics Journal[Internet]. 2005[citado 1 junio 2014]:116(4):984-88 Disponible en:http://pedia-trics.aappublications.org/content/116/4/984.long
7. Kyung Sun Min , Hee Jo Baek, Dong Kyun Han, JuHee You, et al. Wilms’ tumor, aniridia, genitourinary anomalies, and mental retardation (WAGR) syndrome: Successful treatment of the first case with bilateral Wilms’ tumors in Korea. Korean Journal of Pediatrics [Internet]. 2008 [citado 1 junio 2014]:51(12); 1355-58 Disponible en: http://synapse.koreamed.org/Synapse/Data/PDFData/0052KJP/kjp-51-1355.pdf
8. Choudhury N, BhuyanCh, Jagannath J, Deka P, et al. Wilms Tomour with Aniridia: A case Report. Indian Jorunal of medical &Paediatric Oncology [Internet]. 2005 [citado 1 junio 2014] 26(1):43-4. Disponible en: http://medind.nic.in/ias/t05/i1/iast05i1p43.pdf
9. Osorio M, Salgado M, Shoup C. Tumor de Wilms bilateral. Presentación de un caso y revisión bibliográfica. Anales de Radiologia Mexico [Internet]. 2011 [citado 1 junio 2014] 2:121-26. Disponible en: http://www.medigraphic.com/pdfs/anaradmex/arm-2011/arm112i.pdf
10. Almanza L, Arauz AB. Tumor de Wilms Bilateral. Revista Medico Científica [Internet]. 2009[citado 1 junio 2014]:14(2);23-7 Disponible en: http://www.revistamedicocientifica.org/uploads/journals/1/articles/70/submission/ original/70-244-1-SM.pdf
11. Green Daniel M. The treatment od stages I-IV favorable histology Wilms' Tumor. Journal of Clinical Oncology[Internet]. 2004 [citado 1 junio 2014]:22(8);1366-72 Disponible en: http://jco.as-copubs.org/content/22/8/1366.full.pdf

