










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta Médica Boliviana
versión impresa ISSN 1012-2966versión On-line ISSN 2227-3662
Gac Med Bol v.35 n.2 Cochabamba dic. 2012
Caso Clínico
Síndrome de Lesch-Nyhan, reporte de un caso clínico
Lesch-Nyhan syndrome, report of a case
Manuel Antonio Monroy Delgadillo1,2,a, Sthephany Flores Saavedra1,b, Enrique Gonzalo Rojas Salazar1,b
1Facultad de Medicina, Universidad Mayor de San Simón. Cochabamba, Bolivia.2Unidad de Terapia Intensiva Pediátrica, Hospital del Niñ@ Manuel Ascencio Villarroel,Cochabamba, Bolivia.aPediatra-intensivista; bEstudiante de Medicina.*Correspodencia a: Enrique Gonzalo Rojas Salazar.Correo electrónico: enroque.rojas@gmail.com
Recibido el 29 de octubre de 2012.
Aceptado el 23 de noviembre de 2012
Resumen
El síndrome de Lesch Nyhan es una rara patología cuya incidencia es de 1/380 000 de los nacimientos vivos y en cuyas principales manifestaciones aparecen entre los 3 y 6 meses de edad, y se debe a un error congénito del metabolismo de las purinas, que es causado por una mutación del gen estructural ubicado en el cromosoma X, que determina un déficit de la enzima hipoxantina-guanina-fosforribosil-transferasa. Esta enzima no permite la conversión de la hipoxantina a inosina, lo que hace que el nivel de ácido úrico se eleve más de lo normal afectando principalmente al cerebro y riñones; provocando los trastornos que desarrollan este síndrome. Presentamos el caso clínico de un niño de siete años de edad, con diagnóstico de síndrome de Lesch Nyhan, con características de constantes lesiones autoinflingidas como ser mordedura en la lengua, labios, carrillos y dedos de las manos, acompañado de tofos y trastornos óseos en los miembros inferiores consecuencia del ácido úrico elevado. Para este síndrome no existe tratamiento específico, sólo paliativo.
Palabras claves: cromosoma x, cetoacidosis, pediatría, nefrolitiasis, disartria.
Abstract
Lesch Nyhan syndrome is a rare disease whose incidence is 1/380 000 of live births and whose main manifestations appear between 3 and 6 months of age, and is due to an inborn error of purine metabolism, which is caused by a mutation of the structural gene located on the X chromosome, which determines a deficiency of the enzyme hypoxanthine-guanine phosphoribosyl transferase. This enzyme does not allow the conversion of hypoxanthine to inosine, which causes uric acid level rises above normal mainly affecting the brain and kidneys, causing disorders that develop this syndrome. We report a case of a seven year old, diagnosed with Lesch Nyhan syndrome, with features such as self-inflicted injuries consistent bite on the tongue, lips, cheeks and fingers, accompanied by tophi and bone disorders in accordingly lower limbs elevated uric acid. For this syndrome no specific treatment, only palliative..
Keywords: pregnancy, ectopic; gynecological urgency; emergency treatment.
El síndrome de Lesch Nyhan, también conocido como síndrome de automutilación, es un error congénito del metabolismo de las purinas, causado por la mutación de un gen estructural ubicado en el cromosoma X, de herencia recesiva que afecta sólo a los varones, y determina un déficit de la enzima hipoxantina-guanina-fosforribosil-transferasa (HPRT)1. Este trastorno genético está relacionado a dos elementos clínicos que son la sobreproducción de ácido úrico y una serie de trastornos neurológicos graves1,2. Estas manifestaciones son singulares e incluyen distonía de la acción, cetoacidosis, falla de las funciones cognitivas, balismo y un comportamiento de tipo automutilante como ser mordeduras en lengua, labios, carrillos y dedos de las manos1,3.
El síndrome de lesch Nyhan tiene una incidencia aproximada de 1/380 000 de los nacimientos vivos y las manifestaciones comienzan entre los 3 y 6 meses de edad1,4. Esta patología ocurre por igual en los grupos raciales sin ser más frecuente por consanguinidad. En la infancia tardía es clara la hipotonía axial, y la sospecha diagnóstica ocurre generalmente cuando ocurre algún tipo de automutilación compulsiva y disartria2,5.
la deficiencia de HPRT provoca un descenso en la reutilización de bases púricas y una mayor producción en la síntesis de purinas. la manifestación bioquímica inmediata de este defecto enzimático es el aumento de la síntesis de ácido úrico, que se distribuye en cantidades excesivas por todos los fluidos corporales favoreciendo de este modo la precipitación de uratos en el sistema excretor renal, lo que origina cristalu-rias, nefrolitiasis o nefropatías obstructivas3,4. Esta artritis gotosa, suele ser una expresión articular tardía de la deficiencia de HPRT, que se caracteriza por la enorme agresividad que esta presenta y por su aparición en edad juvenil, de depósitos subcutáneos sólidos denominados tofos2,6,7.
Aún no se conoce exactamente la conexión entre el déficit enzimático y las manifestaciones neurológicas características del Síndrome de lesch Nyhan, sin embargo, se estableció que la actividad de la HPRT es más alta en los ganglios basales que en otros tejidos corporales5,8. Diversas fuentes han indicado que los niveles de dopamina, ácido homovanílico, dopades-carboxilasa y, en menor grado, acetilcolina, disminuyen el nivel de serotonina y en cambio los niveles de 5-hidroxiindola-cético aumentan, mientras que el ácido glutámico permanece inalterado7,9. Todas estas fuentes indican que la deficiencia de HPRT condiciona una anomalía cerebral de carácter funcional más que estructural9,10, en la cual, la falta de equilibrio entre las funciones gabérgicas, dopaminérgicas y colinérgi-cas en el sistema extrapiramidal podría ser responsable de las manifestaciones neurológicas características del Síndrome de lesch Nyhan2.
A continuación se presenta el caso de un niño de siete años de edad, con características propias de este síndrome, como ser lesiones autoinflingidas, la presencia de tofos y una serie de trastornos producto del ácido úrico elevado.
Presentación del caso
Paciente de sexo masculino, de siete años de edad, raza mestiza y origen rural, producto del tercer parto de una madre de cuarenta y siete años, quien refiere no haber tenido control prenatal ni antecedentes traumáticos en el parto ni al momento del nacimiento, sin signos de malformación o de disfunción.
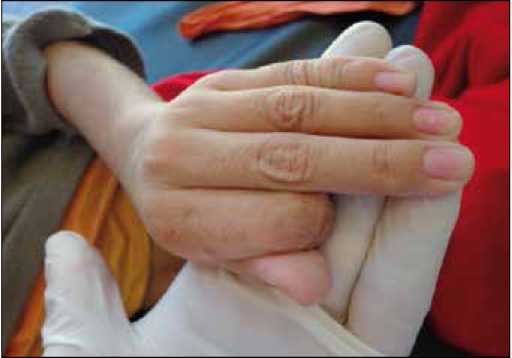
Figura 1. Automutilación de las falanges medial y distal del dedo índice de la mano izquierda.

Figura 2. Presencia de tofos gotosos y deformación articular en miembros inferiores.
El paciente acude al servicio de consulta por presentar un trastorno psicomotor, coreoatetosis, posición de opistótonos, disartria, problemas articulares, tofos gotosos y signos de au-tomutilación, además se evidencia una notoria desnutrición. A nivel neurológico el paciente se encuentra orientado sólo en persona.
La madre relata que al primer año de edad el niño presentó rigidez articular y un trastorno motor, que fue empeorando con los años, junto con una progresiva discapacidad mental, disartria, desórdenes del comportamiento y autoagresividad. Refiere que a los cuatro años, se automutiló las falanges medial y distal del dedo índice de la mano izquierda (figura 1), adquirió la posición de opistótonos, hiperreflexia osteotendi-nosa, y además comenzó a presentar pérdida de apetito y cambios en su orina, haciéndose más oscura y fétida.
A los seis años se evidenciaron tofos gotosos en pies y deformación articular de codos, rodillas y falanges (fig. 2).
A la exploración física, a nivel de la boca se evidencian lesiones en los labios y carrillos (fig. 3), mismas que fueron realizadas de manera paulatina y que actualmente se encuentran
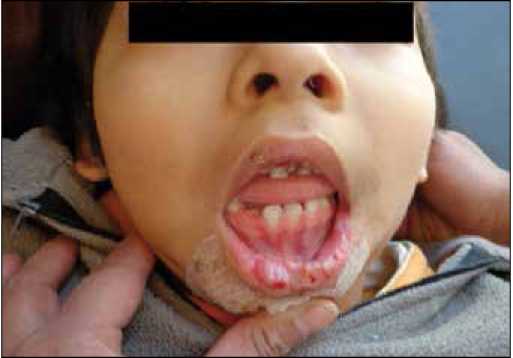
Figura 3. Lesiones de automutilación a nivel de la lengua, labios y carrillos.
protegidas para evitar mayor lesión y proteger la lengua del paciente.
En base a los datos clínicos mencionados anteriormente, se tuvo la sospecha inicial fue que se podría estar ante un paciente con síndrome de Lesch Nyhan; puesto que clínicamente presenta automutilación que es un dato patognomónico de esta patología, acompañado del trastorno neurológico que es otra característica de este síndrome
Posteriormente se realizaron exámenes de laboratorio, entre éstos: Hemograma completo, examen general de orina y ecografía renal, con el fin de confirmar el diagnóstico. En base a las pruebas de sangre se observó una hiperuricemia con un nivel excesivamente elevado (11,1mg/dl) para un niño de su edad, además el hemograma mostró datos compatibles con anemia megaloblástica, característica también de este síndrome. Los demás parámetros evaluados como ser la glicemia, úrea, creatinina, nitrógeno ureico, se encuentraron dentro de los valores normales. En el examen general de orina, se evidenciaron cristales de fosfato amorfo en campo cubierto, flora microbiana en abundante cantidad, nitritos (+), con pH alcalino de 8,5 de aspecto turbio y de olor fétido; además de evaluarse el volumen, color, densidad y urobilinógeno que son normales.
Llamó la antención la excesiva acumulación de cristales de urato monosódico periarticular en tejido celular subcutáneo, sobretodo en superficie dorsal de ambos pies, y el examen físico que muestra la presencia de tofos gotosos, y deformación articular en miembros superiores e inferiores.
En la ecografía renal se evidenciaron riñones eutópicos de tamaño conservado, contornos regulares y definidos, buena diferenciación corticomedular, parénquima conservado, pirámides hiperecogénicas sin sombra acústica, no se observa dilatación pielocalicial. Las dimensiones del riñón derecho son 60 x 38 x 26 mm, con parénquima de 11 mm; el riñón izquierdo mide 70 x 35 x 40 mm, con parénquima de 17 mm. Se observan pirámides renales calcificadas; llegando al diagnóstico de nefrocalcinosis.
Al no existir un tratamiento curativo, se prescribe al paciente Alopurinol, puesto que disminuye la cantidad de ácido úrico formado, eliminando así algunos de los problemas debidos a los depósitos de urato sódico. Se administra una dosis diaria de 5 mg/kg/día de Alopurinol, además de una dieta baja en proteínas, menor a 1,4 g/100 kcal, así como la corrección de la acidosis y del equilibrio hidroelectrolítico con elevado aporte de líquidos.
Dentro el tratamiento neurológico, se utilizó benzodiace-pinas, entre éstas el Diacepam 7mg/Kg/peso, acompañado de la Carbamacepina 10 mg/kg/día administrados en dos veces al día.
La anemia megaloblástica, agravada por su desnutrición, fue tratada con la administración de ácido fólico, vitaminas y cofactores, preferentemente vitamina B12.
Aparte del tratamiento farmacológico se le realizaron una serie de medidas para evitar mayor autolesión como ser dispositivos protectores, mantener los brazos en extensión con férulas, vendar las manos y el uso de protector bucal. luego de haber cumplido un tratamiento de 3 meses, se evidenció una mejora considerable; puesto que los niveles de ácido úrico se redujeron a 7,3 mg/dl, logrando disminuir las lesiones autoin-flingidas, además de controlar la anemia megalobástica.
Discusión
El síndrome de Lesch Nyhan no constituye en sí un problema diagnóstico cuando se presenta en su forma clásica, puesto que algunas de sus manifestaciones como ser la automutila-ción son patognomónicos de esta patología9,10 .
El paciente que describimos, presenta un fenotipo característico de la deficiencia total de la HPRT7,9. Por tal motivo presenta todas las características del síndrome de Lesch Nyhan, al ser un paciente incapaz de caminar y de valerse por sí mismo, con trastorno psicomotor, disartria, con pérdida de apetito, hiperreflexia osteotendinosa, presenta deformación articular de codos, rodillas y falanges, acompañado de tofos en pies y conducta autoagresiva y mutilante8,9. Esta enfermedad debiera investigarse en todo lactante que comience con retraso del crecimiento y desarrollo psicomotor en sus primeros meses, en el que exista el antecedente de aparición precoz de sedimento anaranjado en la orina; especialmente notorio si están deshidratados10.
Diversas fuentes mencionan que la extracción de dientes es la mejor manera de evitar lesiones autoinflingidas, por otro parte se aconseja cubrir la dentadura y/o proteger las partes más vulnerables de ser lesionadas, como ser la lengua, labios, carrillos, y dedos de las manos6,8.
Por el momento, no existe un tratamiento específico para éste síndrome, las opciones se basan sobre todo en controlar las manifestaciones y evitar futuras complicaciones puesto que la mayoría de los pacientes fallecen entre los 10 y 30 años de edad a causa de una insuficiencia renal.
Por tal motivo creemos que el paciente necesita recibir mucha atención y cuidado de sus padres, cumpliendo a caba-lidad el tratamiento para evitar complicaciones y sobre todo velar por mejorar en la calidad de vida del niño, ya que este síndrome continúa siendo un modelo patológico de gran interés, puesto que hasta la fecha no se conoce exactamente su fisiopatología y por tanto no existe un tratamiento específico, solamente se conoce un tratamiento paliativo.
Concluimos que el diagnóstico precoz y la instauración de medidas terapéuticas, nos permiten reducir el grado de complicaciones en niños con deficiencia de esta enzima, logrando de este modo mejorar la calidad de vida del paciente.
Conflictos de interés: los autores declaran no tener conflictos de interés en la publicación de este caso.
Referencias bibliográficas
1. Torres Jiménez R, García García M, García Puig J. Fenotipo variante del síndrome de Lesch-Nyhan. Med Clin (Barc). 2011; 136(2): 63-6. [ Links ]
2. García MG, Torres RJ, Prior C, Puig JG. Normal HPRT, Región Codificadora en la Deficiencia de HPRT Completa y Parcial. Mol Genet Metab 2008; 94: 167-72. [ Links ]
3. Cervantes C, Villagrán U. Paciente con síndrome de Lesch-Nyhan atendido en el Departamento de Estomatología Pediátrica del Hospital Infantil de Tamaulipas. Reporte de caso. Rev Odontol Mex 2008; 12(3): 154-8. [ Links ]
4. Pontón RA. Enfermedades relacionadas con la nutrición. Errores congénitos. Invenio: Revista de investigación académica. 2010; (24): 147-66 [ Links ]
5. Calderón-González R, Calderón-Sepúlveda RF. Tratamiento clínico de la parálisis cerebral. Rev Neurol 2002; 34: 1-6. [ Links ]
6. Flores BL, Espinoza DP, Hernandez H, Escorcia D. Síndrome de Lesch-Nyhan reporte de un caso. 24th International Congress of Pediatrics; 2004 Ago 15-20; Cancún, Mexico.
7. Jinnah HA, Ceballos-Picot I, Torres RJ, Visser JE, Schretlen DJ, Verdu A, et al. Attenuated va-riants of Lesch-Nyhan disease. Brain 2010; 133(Pt 3): 671-89.. [ Links ]
8. Hernández Nieto, L. Síndrome de Lesch-Nyhan. Med Clín (Barc). 1994; 102:699-700. [ Links ]
9. Jinnah HA, Visser JE, Harris JC, Verdu A, La-rovere L, Ceballos-Picot I, et al. Delineation of the motor disorder of Lesch-Nyhan disease. Brain. 2006; 129(Pt 5): 1201-17. [ Links ]
10.- Trucco B. Litiasis urinaria. Manual de Urología Esencial. Universidad Pontificia de Chile. [Online] [Citado 10 Nov 2012]. Disponible en la web: http://escuela.med.puc.cl/publ/manualUrología/ . LitiasisUrinaria.html.

