










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta Médica Boliviana
versión impresa ISSN 1012-2966versión On-line ISSN 2227-3662
Gac Med Bol v.33 n.2 Cochabamba 2010
CASO CLÍNICO
SÍNDROME DE PEUTZ - JEGHERS, A PROPÓSITO DE UN CASO
Syndrome of Peutz - Jeghers a case especially designed
* Marlene Grace Anaya Domínguez, **Alejandra López Terán
*Cirujano pediatra Hospital del Niñ@ "Manuel Ascencio Villarroel"
**Médico Cirujano
Recibido: 30-09-10; Aceptado: 22-10-10
RESUMEN
Se presenta un caso clínico de un niño de 12 años de edad de sexo masculino, Referido al Hospital del Niñ@ Manuel A. Villarroel por un cuadro de obstrucción intestinal baja de 10 días de evolución asociado a manchas melanóticas en piel y mucosas; posterior a múltiples estudios de gabinete y laboratorio, se evidencia masa sólida que ocupa mesogastrio, de 88 mms, que desplaza asas de yeyuno hacia craneal, se le realiza una laparotomía exploratoria, hallándose dos segmentos de intususcepción de intestino delgado y en cada una de ellas, dos masas tumorales en lumen conformando las cabezas de invaginación, se logra la desinvaginación de ambos segmentos resencando dichas masas. El reporte histo-patológico indica Pólipos con núcleos hamartomatosos llegándose al diagnóstico final de Síndrome de Peutz Jeghers.
Palabras clave: Invaginaciones, Pólipos, Hamartomatosos, maculas hiperpigmentadas. Síndrome Peutz Jeghers.
ABSTRACT
We present a case report of a 12 years oíd boy refered to HNMAV with 10 days of lower gastroinstestinal obstruction and mococutaneous melanotic spots, after laboratory and immaging studies was found to have an 88 mm solid rnass in mesogastrium wich displace the jejunum superiorly, exploratory laparotomy was made, the surgical findings were: two tumoral masses at the lumen of the small bowel configuring two heads of invagination, both invagiantions were resolved and both masses were removed. The pathology reports as hamartomatous polyps confirming diagnosis of Peutz - Jeghers Syndrome.
Key words: invaginations, polyps, hamartomatous, melanotic spots, Peutz - Jeghers Síndrome.
INTRODUCCIÓN
El Síndrome de Peutz-Jeghers (SPJ), también conocido como Síndrome hereditario de Poliposis intestinal, es un desorden autosómico dominante caracterizado por el desarrollo de pólipos hamartomatosos benignos en el aparato gastrointestinal y pigmentaciones mucocutáneas características(1,5), asociado a un alto riesgo de desarrollar lesiones de malignidad gastrointestinal y no gastrointestinal.(5)
El gen y la mutación involucrados en la producción de la patología está ubicado en el cromosoma 19p13.3, conocido como STK11/LKB1, codifica una kinasa serina treonina 4-6 (5) con una posible acción supresora de células tumorales, cuya sobreexpresión induce al crecimiento desmedido de las células(12) .
La mutación en dicho gen incrementa el riesgo de desarrollar cáncer de colon y otros tipos de cáncer no gastrointestinales.(3,5)
El síndrome de Peutz-Jeghers es una patología rara, asociado a la Poliposis adenomatosa familiar, su frecuencia es de 1 caso por 60000 a 1 caso por 300000 nacidos vivos en países industrializados. Se describe una frecuencia similar en todas las razas, la edad promedio de presentación es de 23 años en hombres y 26 años en mujeres siendo su distribución similar en ambos sexos.(2,4,5)
El riesgo relativo de morir por cáncer gastrointestinal es 13 veces mayor en pacientes con SPJ. El riesgo de otros tumores (especialmente cáncer de los órganos reproductivos, de mama, páncreas y pulmón) es 9 veces mayor que en la población general.(2,4)
El SPJ se caracteriza por la combinación de lesiones hiper-pigmentadas en piel y mucosas y pólipos gastrointestinales.(2) Estas manchas de melanina, típicas en el 95% de los casos, aparecen como pequeñas máculas de 1 a 5 mm, de color café a azul oscuro de bordes definidos e irregulares, (se localizan en región perineal (94%), manos (74%), mucosa bucal (66%) y pies (62%)).(7)
Aparecen en la infancia temprana, no producen daño,(3) pudiendo disminuir lentamente en la vida adulta y en casos poco frecuentes desaparece completamente.(7)
Las áreas del tracto gastrointestinal que más comúnmente desarrollan pólipos son el intestino delgado, yeyuno e íleon más comúnmente (65-95 %)(7), pero pueden también estar presentes en el colon (60 %) y estómago (50 %)(3,5); el curso clínico característico es el de obstrucción intestinal inducida por los pólipos y sangrado.(4)
Alrededor del 50% de los pacientes desarrolla y muere por cáncer a la edad de 57 años siendo la edad promedio del primer diagnóstico de éste los 42 años. El riesgo acumulado para desarrollar cáncer en pacientes entre 15 y 64 años es de 93%.(2)
Los criterios diagnósticos propuestos por Giardello son:
Poliposis de intestino delgado, con confirmación histopatológica de pólipos hamartomatosos gastrointestinales.
Antecedentes de historia familiar positiva.
Máculas pigmentadas en piel o mucosas.
Tener 2 de los 3 criterios enumerados indica una diagnostico positivo. (1,4)
Entre los estudios de laboratorio, el SPJ puede manifestarse como anemia microcltica hipocrómica por las pérdidas sanguíneas gastrointestinales microscópicas. Se puede detectar antígeno carcinoembrionario (CEA) útil para la monitorización en caso de cáncer.(2) Los estudios de imagen (radiografía baritada de abdomen, ecografía, tomografía computarizada) pueden ayudar a detectar presencia de pólipos y de probables obstrucciones debidos a éstos, mientras que la esofagogastroduodenoscopía y la colonoscopia son útiles para la toma de biopsias al momento del diagnóstico.(2)
El diagnostico definitivo del tipo de pólipos se realiza por el estudio histopatológico de la muestra obtenida.
La cirugía en casos de SPJ incluye la laparotomía y laparoscopía de los problemas gastrointestinales y extragastrointestinales. En casos de hallazgo de pólipos, se realiza una polipectomía y reparar intususcepciones y resecciones intestinales. La laparotomía y resecciones intestinales están indicadas para intususcepciones de pequeño tamaño, obstrucción o sangrado persistente.(2)

PRESENTACIÓN DE CASO
Se presenta a un paciente de sexo masculino de 12 años de edad, procedente de la provincia Chapare, producto de 3er embarazo de padres sin antecedentes patológicos de relevancia, obtenido por parto domiciliario sin complicaciones, a término; recibiendo lactancia materna hasta los 2 años de edad, no refiere antecedentes patológicos de importancia. Hermanos (4), todos menores, sin antecedentes patológicos. La madre presentó 2 abortos espontáneos, desconoce la causa.
Ingresa al servicio de emergencia del HN@MAV referido de otro centro hospitalario de 3er nivel de la misma ciudad, donde es manejado por 10 días, es referido a nuestro centro, con los diagnósticos de:
1. Estreñimiento pertinaz,
2. DNT III secundaria,
3. Sd. De Peutz Jeghers,
4. Anemia microcitica e hipocromía,
5. Soplo sistólico en estudio.
Durante su internación en el primer centro hospitalario se le realiza Rx de abdomen en bipedestación que reporta patrón de miga de pan a nivel de ampolla rectal y sigmoides, Ecografía abdominal que no reporta alteración patológica alguna.
Recibe tratamiento para la constipación con proctoclisis sin resultados favorables.
Laboratorio: Hto: 19,5 % ,
Hb: 5,7 mg/dl,
GB: 5600 mm3,
Plaquetas: 80.4000/ mm3,
VCM: 61,
HCM: 17,7,
Diferencial: neutrof: 68%,
linfocitos: 18%,
monocitos:6%,
eosinofilos:8%,
Proteinemia:
totales: 55,2 g/dl,
Alb: 32,8 g/dl,
Glob: 22,4 g/dl,
Relac A/G: 1,46 g/dl.
Ionograma: K: 3,5 meq/l,
Na: 130 meq/l,
Ca: 0,94 mmosm/l;
Amilasa 35 UI/I,
GPT: 7.0 UI/I,
GOT: 21,2 UI/I.
A su admisión a nuestro centro el paciente se encuentra con FC: 94x', FR: 26x', PA: 100/60; pálido, mucosas pálidas, edema bipalpebral, en boca, máculas confluyentes de color marrón café y azuladas de aproximadamente 0,5 cm de diámetro en labios y cara interna de mejillas; en tórax se evidencia enflaquecimiento, corazón con soplo funcional sistólico en foco tricuspídeo 1/4, pulmones normales, Abdomen, con masa palpable en flanco izquierdo de aproximadamente 11 x7 cm, peristaltismo de lucha, dolor a la palpación generalizada, extremidades con llenado capilar retardado, manchas de color café marrón azuladas en pulpejo de dedos, de aproximadamente 0,2 a 0,3 cm. de diámetro. Ingresa con el diagnóstico presuntivo de: 1. Obstrucción intestinal Baja, 2. Síndrome anémico, 3. DNT II grado. 4. Sd. De Peutz Jeghers
Se le realizan estudios laboratorios y gabinete posteriores a transfusión de dos paquetes globulares:
Hto: 19,8 %, Hb: 6,2 mg/dl, GB: 5,700/mm3,, diferencial: Seg: 68%, Linf: 25%, Mon: 2%, Eos: 1%, Cay: 4%, Plaquetas: 788.000/mm3, TP: 12,7", actividad 92%, INR: 1,06, APTT: 31''. Bioquímica sanguínea con parámetros normales.
Ecografía reporta: Masa ocupante de espacio en mesogastrio, ecogenicidad brillante de 88 x 33 mm, en topografía de mesenterio, en su interior latidos vasculares, compatible con masa sólida de 88 mms en mesenterio, desplazando asas de yeyuno hacia superior.
Se ingresa a quirófano en fecha 16/05, se realiza laparotomía exploratoria, con el diagnóstico pre quirúrgico de masa tumoral abdominal; entre los hallazgos se encuentran: dos masas tumorales que conformaban cabezas de invaginaciones, en lumen de intestino delgado, en yeyuno e íleon, dentro de invaginaciones masivas entre yeyuno e íleon de aproximadamente 1,50 mts. y otra en yeyunoileocólica de 1 mts de longitud. Se logra la desinvaginación exitosa de ambos segmentos, resecando dichas masas.
Diagnósticos postquirúrgicos: 1. Invaginaciones ileocecocólica y yeyunal 2. Síndrome de Peutz Jeghers
Estudio histopatológico reporta: pólipos con núcleos hamartomatosos, compatibles con Sd. Peutz Jeghers.
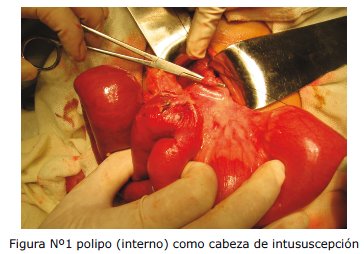
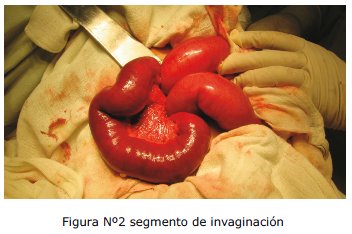
DISCUSIÓN
El caso clínico presentado corresponde a un típico caso de presentación del Síndrome Peutz - Jeghers por presentar las características expuestas coincidentes a la literatura.
El paciente de 12 años de edad, cumple con dos de los tres criterios diagnósticos propuestos por Giardello, los pólipos de intestino delgado con confirmación histopatológica de hamartomas y las máculas hiperpigmentadas en piel y mucosas,(1,4) de tipo macular presentes en la mucosa oral en el 90 % de los pacientes,(4) faltando únicamente los antecedentes familiares para cumplir con el tercer criterio a pesar de haber sido buscados exhaustivamente, lo que se podría explicar por el hecho de que el patrón de herencia es autosómico dominante de penetrancia variable y diversas manifestaciones fenotípicas,(1,2,3) por lo que puede estar o ser subdiagnosticado en sus familiares.
La clínica es característica del cuadro típico de presentación con obstrucción intestinal por pólipos que provocan intususcepción, sangrado intestinal sub diagnosticado, anemia, además de las maculas hiperpigmentadas en piel y mucosas;(2,4,6,7) y acorde a los registros bibliográficos, que indican que la mayoría de los pacientes desarrollan manchas melanóticas durante el primer año de vida, y el primer cuadro de intususcepción entre las edades de 6 a 18 años, el paciente se encuentra entre estos rangos estadísticos característicos. Sin embargo, según datos documentados, la edad promedio del primer diagnóstico es de 23 años. La primera causa de consulta está relacionada con obstrucción intestinal por intususcepción,(1,10) como en el caso actual presentado.
Los resultados de los estudios de laboratorio y gabinete coinciden con los hallazgos documentados para los casos de este síndrome, incluyendo las masas solidas ocupantes en mesogastrio, la obstrucción intestinal, los datos de anemia y los resultados del estudio histopatológico,(2) que indica que la característica de éstos pólipos incluyen afectación del epitelio mucoso, lámina propia y muscular de la mucosa, se trata de grandes pólipos pediculados, con contorno lobulado firme, una gran red arborizada de tejido conectivo y músculo liso que se extiende por todo el pólipo y rodea glándulas normales, ricas en células caliciformes,(9) como indico el resultado del estudio en el caso presentado.
A pesar que la evidencia científica, se concluye que el SPJ no es una enfermedad observada frecuentemente o no es reconocida con facilidad como un síndrome al momento de la consulta ,(10) el manejo del caso presentado fue adecuado, posterior a un diagnóstico oportuno gracias a un alto grado de sospecha de parte del personal capacitado del servicio de cirugía del HN@MAV. A pesar de la ausencia de antecedentes familiares, los cuales fueron buscados con mucho detalle en la familia, la presencia de los otros criterios diagnósticos apoyaban el diagnóstico presuntivo. Se realizaron los laboratorios y estudios de gabinete pertinentes para poder confirmar dicho diagnóstico. La operación realizada (laparotomía exploratoria) que buscaba el origen del cuadro obstructivo, arrojo el último eslabón en el procedimiento diagnóstico al hallar la doble intususcepción en intestino delgado, una vez más, típico del cuadro de presentación y primer diagnóstico del SPJ documentado en la bibliografía. La confirmación final y definitiva lo tiene el estudio histopatológico al dar los resultados de pólipos hamartomatosos.
En cuanto al pronóstico de la enfermedad, interesantes estudios revelaron incremento del riesgo de desarrollo de cáncer en miembros afectados y no afectados, de familias con SPJ, estableciéndose un riesgo de cáncer 18 veces mayor que en la población general, comprometiendo la supervivencia con un riesgo de mortalidad del 40% a los 40 años de vida como promedio,(7) es por tal que el SPJ debe ser diagnosticado con prontitud, ofreciendo un adecuado consejo genético al momento del diagnóstico.(2)
La mayor parte de los pacientes diagnosticados con Síndrome de Peutz-Jeghers desarrollarán alguna forma de neoplasias para la edad de 60 años.(8) Aunque los pólipos hamartomatosos por si mismos no tengan potencial maligno, existe un riesgo creciente de desarrollar carcinoma de páncreas, hígado, pulmones, CA de mama, de los ovarios, útero y testículos(1). Se cree que el cambio histológico maligno ocurre vía cambios adenomatosos en los hamartomas(5). Las neoplasias más comunes los siguientes: esófago, 0.5%; estomago, 29%; intestino delgado, 13%; colon, 39%; páncreas, 36%; pulmones, 15%; testes, 9%; mama, 54%; útero, 9%; ovario, 21%; y cérvix, 10%.(2)
La sobrevida de pacientes con Sd. De Peutz-Jeghers es de 50% a los 57 años de edad, y el riesgo acumulativo de desarrollar una forma de cáncer asociada al síndrome de Peutz-Jeghers entre las edades 15-64 es el 93%(2).
El riesgo de desarrollar cáncer a las edades de 20, 30, 40, 50, 60, y 70 años son del 2%, 5%, 17% 31% 60% y 85% respectivamente. Los casos más comunes de cáncer presentados en estos pacientes son los de origen gastrointestinal, gastroesofágico, intestino delgado, colorectales y pancreáticos; sin embargo, en mujeres, el riesgo de desarrollar cáncer de seno esta significativamente incrementado siendo el 8% y el 31% a las edades de 40 y 60 años respectivamente.(11)
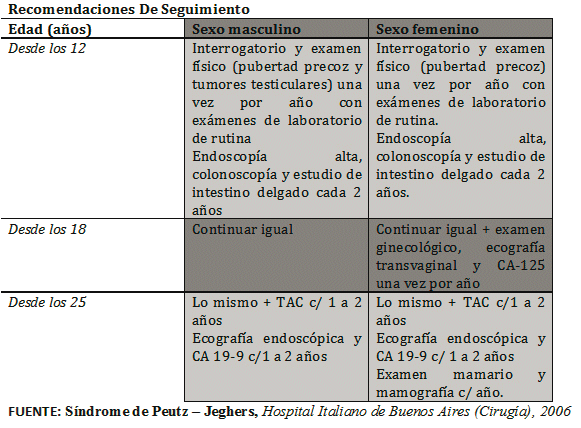
Por las características pronosticas que involucran al SPJ, como ya se expuso, el paciente es sometido a un control periódico postquirúrgico y clínico así mismo se comunico a la familia la posibilidad de presentaciones similares, en el paciente y en sus hermanos. Se plantea que los parientes de primer grado de un caso índice deben ser seguidos anualmente desde el nacimiento, por tanto, un adecuado monitoreo de cáncer intestinal y extraintestinal debe ser implementado (2):
Exámenes físicos anuales.
Hemograma completo anual.
Remoción de pólipos hemorrágicos o de tamaño >5 mm por vía endoscópica.
Vigilancia para el cáncer:
Rx de intestino delgado y colon cada 2 años,
Colonoscopía cada 2 años
Ultrasonografía de páncreas cada año
Ultrasonografía pélvica (mujeres) testicular (hombres) cada año
Mamografía (mujeres) a los 25, 30, 35, y 38 años, después cada 2 años hasta los 50 años, después anualmente o Papanicolaou (Pap) cada año
Una vez realizado el diagnóstico, el manejo debe ser agresivo, con remoción de los pólipos mayores de 5 mm en el estómago y colon, y mayores de 15 mm en el intestino delgado.(6)
Por ser este síndrome parte de las enfermedades documentadas como raras, se recomienda el estudio cuidadoso del mismo para poder determinar con mayor prontitud y exactitud probables casos portadores de esta enfermedad, realizar un diagnostico adecuado, un tratamiento oportuno y un seguimiento continuo.
BIBLIOGRAFÍA
(1 ) Q15831 [STK11JHUMAN de UniProtKB/del Suizo-Prot Kinase 11 del Serine/de la threonine-proteína]. Recuperado encendido 2007-07-21.
(2) Peutz-Jeghers Syndrome: overview, Sandeep Mukherjee, Andrea Duchini, John M Carethers; eMedicine Specialtie; Updated: Apr 9, 2009.
(3) Síndrome de Peutz - Jeghers, Hospital Italiano de Buenos Aires (Cirugía), 2006 [ Links ]
(4) Síndrome De Peutz-Jeghers. Reporte De Un Caso, M. Homan, Z. Dolenc Strazary R. Orel, Acta Dermatoven APAVol. 14, 2005, N° 1.
(5) Síndrome De Peutz-Jeghers Y Adenocarcinoma De Colon, J. F. Pinto Sánchez, S. Rebaza Vásquez, S. Muñoz Mendoza, V. Maco Cárdenas, Hospital Central de la Sanidad de la PNP, REV. GASTROENTEROL. PERÚ 2004; 24: 363-367
(6) Una familia con síndrome de Peutz-Jeghers, Myriam Ferreiro C, Paul Harris D., Francisco Larraín B., Ignacio Duarte G., Gabriela Repetto L, Rev. chil. Pediatr. V.71 n3. Santiago, mayo 2000
(7) Síndrome de Peutz-Jeghers. A propósito de un reporte familiar en el Hospital Arzobispo Loayza; Adelina Lozano, Víctor Valencia y cols.; Revista de Gastroenterología del Perú - Volumen 16, N°l 1996
(8) Peutz-Jeghers syndrome; From Wikipedia [internet] [ Links ]
(9) Patología estructural y funcional, Robbins y Cotran, Pags. 863, 865, 7ma edición, Elsevier, 2005. [ Links ]
(10) Incidence of Peutz-Jeghers syndrome, Data derived from a survey of hospitals , Austin H. Kutscher, Edward V. Zegarelli, Robin M. Rankow and Terry W. Slaughter; Digestive Diseases and Sciences Volume 5, Number 6, 576-577 [ Links ]
(11) Frequency and spectrum of cancers in the Peutz-Jeghers síndrome, Hearle N, Schumacher V, Menko FH, Olschwang S, et cols., Clin Cáncer Res. 2006 May 15;12(10):3209-15. [ Links ]

