








Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkGaceta Médica Boliviana
versão impressa ISSN 1012-2966versão On-line ISSN 2227-3662
Gac Med Bol v.33 n.1 Cochabamba 2010
CASO CLÍNICO
SÍNDROME DE PIERRE ROBÍN
Pierre Robin Syndrome
*Eduardo Suárez Barrientos, **Dánova Andrea López Fajerstein, ***Heydi Sanz Arrazóla
*Servicio de Medicina Hospital del Niñ@ Manuel Ascencio Villarroel.
**Estudiante de 5to año de Medicina, UMSS.
***Neurologa Pediatra Hospital del Niño Manuel Asencio Villarroel
Recibido: 01-12-10; Aceptado: 16-04-10
RESUMEN
Se presenta el caso de una lactante menor de 5 meses de edad, internada en el Servicio de Medicina Hospital del Niñ@ Manuel Ascencio Villarroel con el diagnóstico de Síndrome de Pierre Robin y diversas malformaciones concomitantes: mucocele lingual, colpocefalia, hipoplasia de fosa posterior, nistagmus, paraparesia espástica, retraso del desarrollo, displasia bilateral de cadera, 6o ortejo en miembro inferior derecho, desnutrición crónica y reiterativos cuadros de neumonía intersticial.
Palabras claves: Síndrome de Pierre Robin, Glosoptisis, Micrognatia, Retrognatia, Obstrucción de la vía aérea.
ABSTRACT
A case of an infant under 5 months of age, admitted to the Medicine Service in the Hospital Manuel Ascencio Villarroel, diagnosed with Pierre Robin Syndrome and various malformations concomitant lingual mucocele, colpocephaly, hypoplastic posterior fossa, nystagmus, spastic paraparesis, developmental delay, bilateral hip dysplasia, 6th toe in the right leg, chronic malnutrition and repetitive tables interstitial pneumonía.
KEYWORDS: Pierre Robin Syndrome, Glosoptisis, micrognathia, retrognathia, airway obstruction
INTRODUCCIÓN
Este síndrome fue descrito el año 1923 por Pierre Robin como una dificultad respiratoria asociada a glosoptisis e hipoplasia mandibular. Hoy en día el síndrome es caracterizado por retrognatia o micrognatia, glosoptisis y obstrucción de la vía aérea.1
El síndrome de Pierre Robin debe su génesis a un desarrollo defectuoso del primer arco braquial que se encarga de la formación de los ojos, oídos, mandíbula y paladar, por tanto las anomalías se desarrollan en dichos órganos.
Existe una migración insuficiente de células de la cresta neural hacia el primer arco durante la 4a semana de desarrollo embrionario. Se genera una detención del desarrollo de la mandíbula o "micrognatia", la falta de espacio empuja la lengua hacia el paladar, interponiéndose en el cierre de éste dejando una fisura de paladar en forma de "U". La micrognatia puede provocar la caída de la lengua, "glosoptosis", hacia la faringe, generando una obstrucción de la vía aérea. En los pacientes con micrognatia o retrognatia, el mentón es desplazado posteriormente causando la caída de la lengua hacia la región posterior de la pared faríngea. Esto genera la obstrucción de la vía aérea durante la inspiración. El llanto en los niños tiende a mantener la vía aérea abierta, en cambio al caer dormidos puede generarse obstrucción de la misma. La dificultad para la alimentación en estos niños es muy severa, este hecho puede suscitar una secuencia de eventos: glosoptisis, obstrucción de la vía aérea, llanto, disminución de la ingesta oral y por tanto retraso del crecimiento y desarrollo. Si este círculo vicioso no es tratado, puede llevar al agotamiento, falla cardiaca y finalmente la muerte.1
El tratamiento de este síndrome puede ser dividido en una terapia conservadora versus intervenciones quirúrgicas. La mayoría de estos infantes pueden ser manejados colocándolos en posición prono hasta que exista un adecuado crecimiento de la mandíbula. Esto genera que la mandíbula y la lengua caigan hacia delante dejando libre la vía aérea. Si este tratamiento fracasa puede considerarse una glosopexia, traqueostomía o expansión mandibular también llamada distracción ósea.2,4
El manejo precoz y efectivo de este problema es determinante, ya que la incoordinación de los mecanismos de succión y deglución acompañados de un alto gasto energético destinado a mantener una adecuada ventilación respiratoria, comprometen el estado nutricional y la calidad de vida del paciente.2,4,9
PRESENTACIÓN DE CASO
Paciente de sexo femenino producto de madre primigesta sin antecedentes de relevancia identificados. Nace el 31 de Agosto del 2008 mediante cesárea, desconociéndose otros detalles sobre el nacimiento.
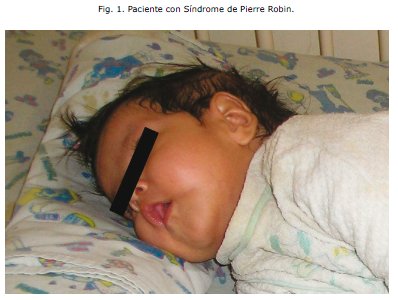
En fecha 22 de Noviembre del mismo año, ingresa al Hospital del Niñ@ Manuel Ascencio Villarroel, a los 5 meses de edad con severa dificultad para la succión de leche materna y con accesos de tos intermitentes. En el Servicio de emergencias, al examen físico destacaban: microcefalia, con un perímetro cefálico de 35 cm, la presencia de mucocele lingual de 1cm x 1cm de diámetro, pabellones auriculares de implantación baja, roncus en ambos campos pulmonares, la presencia de un 6o ortejo en miembro inferior, además de un pobre desarrollo pondoestatural.
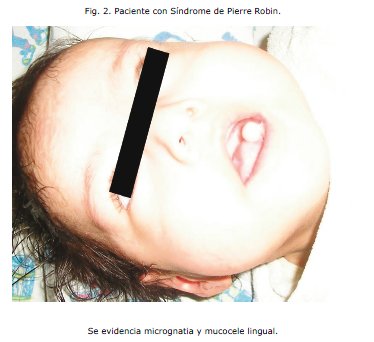
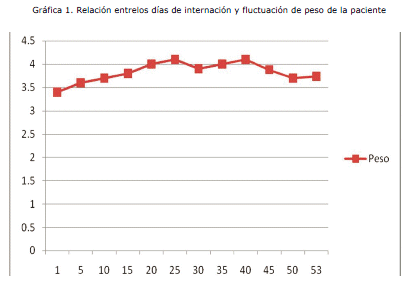
Al observar la severidad del cuadro es derivada al Servicio de cuidados intermedios de pediatría (Medicina I), y una vez estabilizado el cuadro es referido el 25 de febrero al Centro de Rehabilitación Integral Nutricional (CRIN), con un peso de 3,82 kg y una talla de 55cm. Durante su estancia en esta sala manifiesta nuevamente un cuadro respiratorio que corresponde a una neumonía atípica por lo que es referido nuevamente al servicio de cuidados intermedios para su manejo (Medicina II). Llamó la atención en este servicio el hecho de que la paciente tenía una curva de crecimiento estacionaria aún brindándole los requerimientos calóricos y proteicos del soporte nutricional im-plementado; no evidenciaba mejoría en el peso aún con un incremento periódico en la ingesta. De hecho la gráfica sobre el peso no tenía fluctuaciones relevantes incluso en el 53° día de internación, manifestando una Desnutrición Crónica, este hecho generó preocupación en el equipo médico y condujo a plantear la sospecha de alguna patología de base que impedía el crecimiento y desarrollo normal de la paciente. Se realizó un minucioso protocolo de estudio y se detectaron las siguientes anomalías: parálisis cerebral infantil, retardo del desarrollo neurológico y psicomotor, paladar gival, malformación mandibular, nistagmus horizontal, probable amaurosis central, aumento del 1o y 2o ruido cardiacos a predominio del segundo, desdoblamiento fijo y síndrome regurgitativo, ductus arterioso persistente acompañado también de displasia bilateral de cadera.
En base a lo señalado, la evaluación por Medicina II añade la sospecha clínica de Síndrome de Pierre Robin consistente en: Glosoptisis, Micrognatia, Fisura palatina y Cardiopatía, coincidentes con los hallazgos presentes en la paciente.
Y se decide un manejo multidisciplinario (cardiología, neurología, fisioterapia y oftalmología) para abordar el problema. El soporte nutricional fue adaptado al estrés adicional de la patología asociada como se recomienda en el manejo de la desnutrición secundaria y fue referido nuevamente al centro de rehabilitación nutricional.
Exámenes paraclínicos
Biometría hemática, química sanguínea, ionograma y estudio coproparasitológico dentro de parámetros normales. Estudios de imagen comentados a continuación:
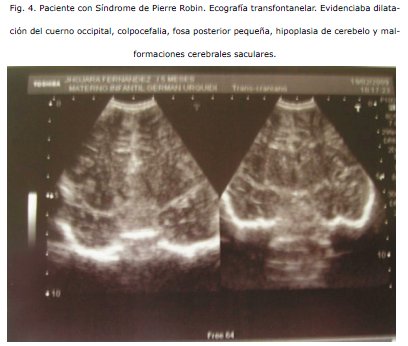
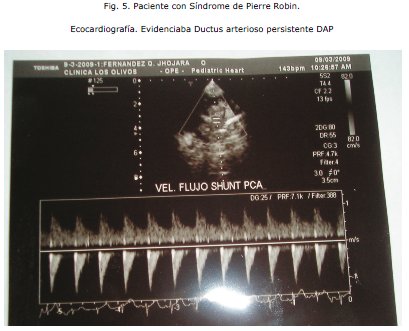
COMENTARIO
Etiología y Patogénesis
Frecuencia
Este es un heterogéneo defecto de nacimiento que tiene una prevalencia de aproximadamente 1 en 8500 nacidos vivos. Con igual razón entre niños y niñas, excepto en la forma ligada al Cromosoma X. 3
Etiología
Es posible que se deba a una herencia Autosómica recesiva. Una variante ligada al Cromosoma X-a sido reportada incluyendo malformaciones cardiacas.1
Patogénesis
Existen tres teorías fisiopatológicas para explicar la ocurrencia de la Secuencia de Pierre Robin:2
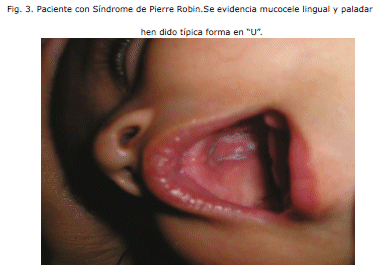
- La teoría mecánica: Esta es la teoría más aceptada. El evento inicial, hipoplasia mandibular, ocurre entre la 7a y 11a semana de gestación. Esto mantiene a la lengua alta en la cavidad oral, causando una hendidura palatina pre viniendo que se cierren los pilares del paladar. Esta teoría explica la clásica hendidura en forma de "U" invertida y la ausencia de un labio hendido asociado. El Oligohidramnios podría desempeñar un papel en la etiología ya que la ca rencia del líquido amniótico podía causar la deformación de la barbilla y del impacto subsecuente de la lengua entre los pilares del paladar.2
- La teoría de la maduración neurológica: Un retardo en la maduración neurológica se ha observado en la electromiografía de la musculatura de la lengua, de los pilares faríngeos, y del paladar, ya que tiene un retardo en la conducción del nervio hipogloso. La corrección espontánea de la mayoría de casos con edad apoya esta teoría.2
- La teoría de desneuralización romboencefálica: En esta teoría, el motor y la organización reguladora del romboencéfalo se relaciona con un problema grave de la ontogénesis.6
Manifestaciones Otorrinolaringológicas
La micrognatia está presente en la mayoría de los casos (91.7%). Es caracterizada por la retracción del arco dental inferior 10-12 milímetros detrás del arco superior. La mandíbula tiene un pequeño cuerpo, un ángulo cordial obtuso, y un cóndilo localizado posteriormente.3
El crecimiento de la mandíbula se alcanza durante el primer año; sin embargo, la resolución de la hipoplasia mandibular en el niño logra un perfil normal aproximadamente a los 5-6 años de edad. Se observa Glosoptosis en 70-85% de los casos divulgados. La Macroglosia y anquiloglosia son resultados relativamente raros, conocidos en un 10-15% de casos divulgados. La combinación de micrognatia y de glosoptosis puede causar dificultad respiratoria y severa de alimentación en el recién nacido. La obstrucción respiratoria de la apnea del sueño puede también ocurrir.
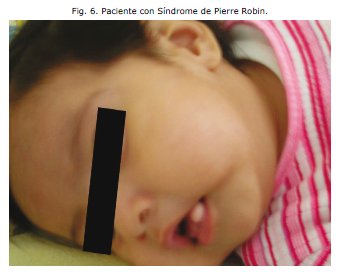
Se evidencia micrognatia, glosoptisis, mucocele lingual Y orejas de implantación baja.3
En series reportadas, el predominio del paladar hendido varía de 14-91%. Puede afectar al paladar suave y duro y es generalmente en forma de "U" (el 80%) o en forma de V. De vez en cuando, se puede presentar como campanilla bífida o doble o como hendidura submucosa oculta. 3
La anomalía ótica más común es la otitis media, en un 80%, seguido por anomalías auriculares en el 75% de casos. La pérdida de oído, sobre todo conductora, ocurre en el 60% de pacientes, mientras que la atresia externa del canal auditivo ocurre en el solamente 5% de los pacientes. Se demuestra inadecuada neumatización del hueso temporal de las cavidades mastoideas en muchos pacientes con secuencia de Pierre Robin.
Las deformidades nasales son infrecuentes y consisten sobre todo en anomalías de la raíz nasal. Las malformaciones dentales ocurren en una mitad de casos. La larin-gomalacia ocurre en aproximadamente 10-15% de pacientes con secuencia de Pierre Robin. El reflujo gastro-esofágico y la esofagitis también se han descrito. Los defectos en el lenguaje ocurren con frecuencia en pacientes con secuencia de Pierre Robin. 3
Manifestaciones Sistémicas
Las anomalías sistémicas se documentan generalmente en 10-85% de los casos reportados. Las anomalías oculares se han reportado en 10-30% de pacientes.5 La frecuencia más alta se observa generalmente cuando se consulta a un oftalmólogo. Las lesiones siguientes ocurren por orden decreciente de frecuencia: hipermetropía, miopía, astigmatismo, esclerosis de córnea, y estenosis nasolacrimal del conducto.
Los resultados cardiovasculares tales como murmullos benignos, estenosis pulmonar, Conducto arterioso persistente, Agujero oval persistente, defecto septal atrial e hipertensión pulmonar todos se han documentado su prevalencia varía en la literatura de 5-58%.5,6,7
Las anomalías que implican el sistema musculoesqueletico son las anomalías sistemicas más frecuentes (conocidas en 70-80% de casos). Incluyen sindactilia, falanges displásticas, polidactilia, clindactilia, empalmes hiperextensibles y oligodactilia en los miembros superiores. En las extremidades inferiores, se han reportado las anomalías del pie, las malformaciones femorales (varu o valgus de la cadera, fémur corto), las anomalías de la cadera (contracciones de la flexión, dislocación congénita), las anomalías de la rodilla (valgus, sincondrosis de la rodilla), y las anormalidades tibiales. Las deformidades de la columna vertebral incluyen escoliosis, cifosis, lordosis, displasia vertebral, agenesia sacra. 6
Las manifestaciones a nivel neurológico incluyen retardo en el lenguaje, epilepsia, retardo del desarrollo psicomotor, hipotonía, e hidrocefalia pueden ocurrir. La incidencia de las manifestaciones del SNC es alrededor del 50%.5
Los defectos genitourinarios pueden incluyen criptorquidea (el 25%), el hidronefrosis (el 15%), y el hidrocele (el 10%).
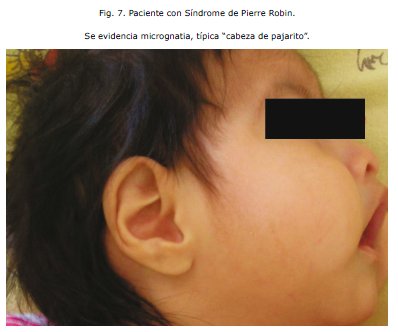
El caso reportado corresponde a típicos hallazgos que identifican la Secuencia de Pierre Robin: micrognatia, fisura palatina, glosoptisis y dificultad respiratoria por obstrucción de la vía aérea; todas estas alteraciones fueron asociadas al ver que la paciente tenía una curva de crecimiento estacionaria aún con los requerimientos calóricos y proteicos del soporte nutricional. Este hecho generó preocupación en el equipo médico y condujo a plantear la sospecha diagnóstica de Síndrome de Pierre Robin, por la severa dificultad para la alimentación del Recién Nacido y la falta de ganancia de peso y talla; puesto que las malformaciones que presenta dicho síndrome evitan una adecuada alimentación del paciente. El caso clínico citado presenta también la típica hendidura palatina en forma de "U" que caracteriza a la patología y no así un paladar hendido en forma de "V" que sería el más usual en cualquier otro síndrome. Las malformaciones cardiacas frecuentemente presentes en este cuadro, se manifiestan por un Conducto Arterioso Permeable en nuestro reporte. No presenta malformaciones auditivas ni nasales, pero si existen anomalías visuales con nistagmus y posible amaurosis, en cuanto a las manifestaciones sistemicas los trastornos músculo esqueléticos son un patrón importante y en este caso se evidenció la presencia de displasia bilateral de caderas y un 6o ortejo en MID, mucocele lingual, colpocefa-lia, hipoplasia de fosa posterior, parálisis cerebral infantil, retardo del desarrollo neurológico y psicomotor. Un dato sobresaliente es que la paciente presentó reiterativos cuadros de neumonía probablemente secunadios a un síndrome aspirativo como es de esperar en estos casos.
Al tener compromiso en el primer arco branquial se realizó el diagnóstico diferencial con diversos cuadros que afectan dicha región como el síndrome de Treacher Collins. Al ser los arcos branquiales dependientes de la migración de células de la cresta neural para su desarrollo, es probable que las malformaciones cardiacas y neurológicas de la paciente también se deban a un defecto en la migración de las células de la cresta neural encargada de la formación de los mismos. Los síndromes y las condiciones asociados incluyen síndrome Stickler, la trisomía llq, trisomía 18, el síndrome velocardiofacial (de Shprintzen), el síndrome de la delección 4q, la artropatía reumatoide, el síndrome de Móbius, y la asociación CHARGE.6
El soporte nutricional inicial en la paciente fue establecido como para el tratamiento de una desnutrición primaria. Las características referidas en este caso evidencian una desnutrición secundaria lo que implica un estrés adicional incrementando sus demandas energéticas en el contexto de una paciente cardiópata.8 Estos niños tienen historia de pobre ganancia de peso y falla para progresar, por lo que frecuentemente requieren concentración de la fórmula y/ o suplementación para darle al niño una mayor ingesta de calorías y nutrientes, conducta que fue tomada con nuestra paciente.
En general los niños con este síndrome tienen un pronóstico bueno, ya que el desarrollo mandibular se alcanza hasta el año de edad, a veces sin necesidad de intervenciones quirúrgicas sobre la mandíbula, limitándose solamente a reparar la fisura palatina.9,10
RECOMENDACIONES
Es importante plantear diagnósticos sindrómicos en niños con múltiples malformaciones de modo que ayuden a la identificación de la causa y manejo del paciente. La presencia de 3 malformaciones menores sugiere la posibilidad de alguna malformación mayor, por tanto debemos estar atentos al diagnóstico diferencial. Si estamos ante un paciente con desnutrición y éste no recupera el estado nutri-cional con la alimentación que le suplementamos indica que el déficit no es primario sino secundario, por lo que se debe buscar una causa secundaria de este crecimiento estacionario y actuar en consecuencia
BIBLIOGRAFÍA
1. MOROVIC I, Carmen Gloria. Manejo actual en síndrome de Pierre Robin. Rev. chil. pediatr. [online]. 2004, vol.75, n.1 ISSN 0370-4106. SciELO Chile. [ Links ]
2. MARQUES, Ilza L. et al. Seqüéncia de Robin: protocolo único de tratamento. J. Pediatr. (Rio J.) [online]. 2005, vol.81, n.1 ISSN 0021-7557. : SciELO Brasil. [ Links ]
3. CASTRO, Aliñe Maria Alencar de y VASCONCELOS, Maria Helena Ferreira. Avaliacao da influencia do tipo facial nos tamanhos dos espacos aéreos nasofaríngeo e bucofaríngeo. Rev. Dent. Press Ortodon. Ortop. Facial [online]. 2008, vol.13, n.6 ISSN 1415-5419.:SciELBrasil. [ Links ]
4. MOROVIC I, C. Gloria y MONASTERIO A, Luís. Alargamiento mandibular mediante tracción en pacientes con síndrome de Pierre Robin. Rev. chil. pediatr. [online]. 1996, vol.67, n.6 ISSN 0370-4106.: SciELO Chile. [ Links ]
5. DENNISON WM.: The Pierre Robin Syndrome, PedMtrics 36: 336, 1965.
6. FREEMAN M.K., MANNERS J.M.: Cor Pulmonale and the Pierre Robin anomaly, Anaesthesia 35: 282,1980.
7. COGSWELL J.J., EASTON DM.: Cor Pulmonale in the Pierre Robin Syndrome, Arch. Dis. Child. 49: 905,1974.
8. GOLDBERG M.H., ECKBLOM R.H.: The treatment of the Pierre Robin Syndrome, Pediatrics 30: 450,1962.
9. DOUGLAS B.: THE treatment of micrognatia associate with obstruction by a plástic procedure, Plast.Reconstr. Surg. 1: 300, 1946.
10. HEAF D.P, HELMS PJ, DINWIDDIE R., MATTHEW DJ.:Nasopharyngeal airways in Pierre Robin Syndrome,The J. of Pediatr. 100: 698, 1982.
Este artículo fue revisado bajo las Normas Internacionales de Index Médicas