










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO Compartir

 Permalink
PermalinkGaceta Médica Boliviana
versión impresa ISSN 1012-2966versión On-line ISSN 2227-3662
Gac Med Bol v.30 n.1 Cochabamba 2007
CASO CLINICO
SÍNDROME DE APERT (ACROCEFALOSINDACTILIA)
* Alfredo Villarroel Goytia,** Erwin Hochstatter Arduz,** Roxana Claustro
* Ginecologo Obstetra de la CNS Hospital Obrero No.2 Docente de Ginecología y Obstetricia de la Facultad de Medicina - UMSS
** Residente III de Ginecología y Obstetricia . Hospital Obrero No.2 de la Caja Nacional de Salud
RESUMEN
Se presenta el reporte de un caso de recién nacido con síndrome de Apert, cuadro de baja frecuencia en nuestro servicio. Se hace una revisión de la literatura constatándose que es una enfermedad con un componente genético y que corresponde con algunas cromosomopatías que presentan deformidades craneofaciales importantes. Las características más relevantes de este síndrome radican, en la fusión de algunos huesos del cráneo, manos y pies, dándole al paciente un aspecto característico. Su tratamiento entra en el orden multidisciplinario ya que son múltiples las deformidades en esta enfermedad.
Palabras clave: Acrocefalia, sindactilia, Síndrome de Apert.
ABSTRACT
We report an case of newborn with Apert's syndrome. A medical literature revision was made concluding that Apert's syndrome is a congenital disease, associated with some chromosomopathies that express important cranial and facial deformities. The most important characteristics of this syndrome are fussions in some bones of the crane, hands and feet; giving the patient a characteristic aspect. It's Treatment must be multidisciplinar}' because of the multiple deformities presented in this syndrome.
KEY WORDS: Acrocephaly, syndactyly, Syndrome of Apert.
INTRODUCCIÓN
El síndrome de Apert o acrocéfalo-sindactilia es una enfermedad genética que puede ser hereditaria, de rasgo autosómico dominante o que también puede presentarse sin que existan antecedentes familiares conocidos y deberse a mutaciones esporádicas del gen F6FR2 del cromosoma numero 10 con carácter no recesivo, que codifica una proteína llamada factor Receptor 2 de crecimiento fibroblástico, y que cuando se produce la mutación, causa la fusión prematura de las suturas craneales. Se han descrito mutaciones en otros genes como el P253R, relacionado con la sindactilia y el S252W, con el paladar hendido.
La frecuencia de aparición es de aproximadamente 1 caso de cada 160.000 nacimientos con una distribución equitativa entre el sexo masculino y femenino. Los asiáticos tienen el predominio más alto (22.3 por millón de nacidos vivos), y los hispanos tienen el predominio más bajo (7.6 por millón de nacidos vivos)2. Algunos estudios han demostrado que la aparición de esta patología es mas frecuente entre los hijos de parejas de edad avanzada.
Se caracteriza por presentar malformaciones específicas en el cráneo, tercio medio de la cara, manos y pies. El cráneo se fusiona prematuramente impidiendo su crecimiento normal, el tercio medio de la cara está hipoplásico dando un aspecto de hundimiento del mismo, y las manos y pies presentan una fusión entre los dedos, la cual puede variar en cuanto a involucrar solamente los tejidos blandos o extenderse también a los huesos (Kaplan. 1991).
El síndrome de Apert presenta una serie de signos clínicos diseminados a lo largo de toda la arquitectura corporal del individuo que lo padece, siendo los más importantes los siguientes:
a) En el Cráneo: Craneosinostosis, (obliteración temprana de las suturas craneales) acrocefalia, (cabeza en forma de cono) turribraquicefalia. (diámetro antero-posterior del cráneo disminuido) y aplanamiento de la frente y del occipucio. (Gorlin, 1976; Sedano, 1977)
b) En la Cara: Hipoplasia A-P del tercio medio de la cara, hipoplasia del reborde orbitario dando la impresión de proptosis de los globos oculares, interrupción en la continuidad de las cejas, asimetría facial, puente nasal hundido, hipertelorismo, mandíbula prominente como efecto de la hipoplasia del tercio medio, fisuras palpebrales antimon-goloides, pabellón auricular grande y generalmente en localización baja.
c) En los Maxilares: Maxilar superior con paladar ojival, pudiendo presentar fisura palatina o úvula bífida, arco maxilar en forma de V con múltiples apiñamientos dentales, hiperplasia gingival generalizada, la mandíbula aparece en relación Clase III con respecto al maxilar superior. También se ha reportado alteraciones en el patrón de erupción de los dientes (Kaloust, 1997)
d) En las Extremidades: Las manos y los pies presentan, fusión generalmente de los dedos índice, medio y anular, y segundo, tercero y cuarto dígito respectivamente, presentan sindactilia. Puede existir la presencia de una sola uña grande entre los dedos fusionados lo cual da el aspecto característico de "cuchara de sopa". La unión puede estar confinada solamente a los tejidos blandos o por el contrario involucrar también al hueso en cuyo caso se presenta una fusión parcial o total de los dedos. Las extremidades superiores se encuentran acortadas con aplasia o anquilosis de algunas articulaciones, especialmente de los hombros, codos y cadera (Gorlin, 1976; Sedano. 1977).
e) Aspecto Neurológico: Puede existir una inteligencia normal o presentar varios grados de retardo mental, siendo más común el retardo moderado, sin embargo, se han reportado casos de individuos con una inteligencia normal. También podemos encontrar en algunos casos hidrocefalia con la consecuente alteración de la presión intracraneal. La razón aparente por la cual éste síndrome cursa con problemas de retardo, son las malformaciones que se han encontrado en el cuerpo calloso del cerebro y otras estructuras adyacentes (Cohen, 1990). El desarrollo intelectual de los pacientes con Apert es el relacionado con el medio donde se desenvuelven a diario.
f) En la Piel: Se presenta hiperhidrosis generalizada y acné vulgaris severo en la cara, pecho, espalda y miembros superiores, también llama la atención el exceso de arrugas en la piel que cubre la frente lo cual es debido a la hipoplasia ósea presente en el hueso frontal. (Cohen y col, 1995)
g) Aspecto Sistémico: Algunos afecciones que se relacionan son: defectos cardiovasculares, atresia pulmonar, ducto arterial permanente, fístula traqueoesofágica, estenosis pilórica, riñones poliquísticos, infecciones otológicas y apnea del sueño (Kaplan, 1991). La flexibilidad de columna cervical se encuentra seriamente afectada.
Debido a la fusión que presentan estos pacientes entre las vértebras cervicales C5-C6, el estudio radiográfico de la columna cervical es muy importante, especialmente en la evaluación anestésica preoperatoria ya que la flexibilidad del cuello necesaria para la intubación endotraqueal se encuentra seriamente afectada, haciendo de éste, un acto complicado y difícil. (Kreiborg, 1992).
Tratamiento
El tratamiento de los pacientes con síndrome de Apert debe ser enfocado desde un punto de vista integral y requiere del esfuerzo combinado de profesionales de diversa indole.
La evaluación de estos pacientes comienza desde el nacimiento y la infancia, donde el pediatra, el neurocirujano, el oftalmólogo, el otorrinolaringólogo, el odontopediatra, el cirujano craneofacial. el neuroradiólogo, el psicólogo y el trabajador social son esenciales. Luego durante las etapas de la adolescencia y juventud temprana se suman al equipo de tratamiento, el ortodoncista, el odontólogo general, el terapista de lenguaje y el cirujano maxilofacial. (McCarthy y col, 1976)
El tratamiento quirúrgico debe ser precoz, antes de los 6 meses de edad y va dirigido a descomprimir el espacio intracraneal y evitar el edema papilar. El avance del tercio medio facial que incrementa el volumen intra craneal e intra orbitario mejora el flujo aéreo nasal y la función respiratoria y permitir el desarrollo normal de las distintas áreas cerebrales.
El tratamiento quirúrgico debe orientarse también a mejorar no sólo el aspecto físico del niño, sino, además, las diversas alteraciones funcionales, sobre todo en cara y manos, aunque estas cirugías se realizan mas tarde, alrededor de los 6 años. Esta selección cuidadosa de los tiempos quirúrgicos redunda únicamente en los buenos resultados estéticos y funcionales que se persiguen a largo plazo.
Pronóstico
El pronóstico depende en gran parte de la edad en la operación. Las innovaciones en cirugía craneofacial han permitido a niños con el síndrome de Apert alcanzar su capacidad adecuada maximizando sus oportunidades para el crecimiento intelectual, la capacidad física, y la aceptación social; sin embargo, el tratamiento quirúrgico temprano de la craneosinostosis puede no alterar el resultado intelectual. El pronóstico depende de malformaciones asociadas del cerebro y la calidad del ambiente familiar es otro factor implicado en el logro intelectual.
Diagnósticos diferenciales
Una característica que diferencia al síndrome de Apert de los otros síndromes del grupo de las craneosinostosis como los síndromes de Crouzon, Fraccaro y con el síndrome 18q heredado y con otros que cursan con craneosinostosis, como los de Carpenter, de Saethre-Chotzen y de Pfeiffer, es la presencia de fusión de los dedos, lo cual se conoce como sindactilia.
CASO CLINICO
Paciente de 29 años, segundigesta, que acude al Servicio de Gineco - Obstetria del Hospital Obrero No 2; referida del policlínico No. 32 de la Caja Nacional de Salud con el diagnostico de embarazo de 32 semanas por U.P.M.
Antecedentes personales : No refiere ninguno significativo
Antecedentes familiares: Padres aparentemente sanos, esposo aparentemente sano, hija de 14 años con parálisis cerebral residual.
Antecedentes gineco-obstetricos: G2 P1 A0 C0. G1: parto eutócico hospitalario hace 14 anos, con secuela de parálisis cerebral secundaria a sufrimiento fetal agudo severo.
Historia del embarazo:
Realizó controles prenatales en Policlínico No 32, referida al servicio de Gineco-obstetricia de Hospital Obrero No 2, en fecha 16.04.06.
Se realizan exámenes complementarios de rutina:
10-10-05:
Hemograma normal, grupo O Rh (+); glicemia 101mg/dl, Chagas HAI: 1/32; ELISA: positivo; VDRL negativo; To-xoplasmosis negativa (IgG , IgM).
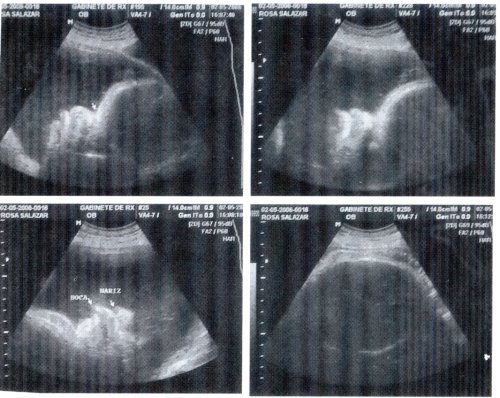
Fig. 1: Ecografía del 26 de abril de 2006: tiempo gestacional de 36.5 semanas
Parcial de orina: compatible con infección urinaria Ecografía: Embarazo de 7,1 semanas.
26-04-06:
Ecografía obstétrica: Embarazo de 36,5 semanas +/- 3 semanas por longitud femoral; polihidramnios, probable microcefalia con hidrocefalia leve. DBP circunferencia craneana en p5 y p<5 respectivamente, con ventriculomegalia de 14 mm bilateral a nivel de atrio. Deformidad del rostro por frente prominente e hipoplasia nasal.
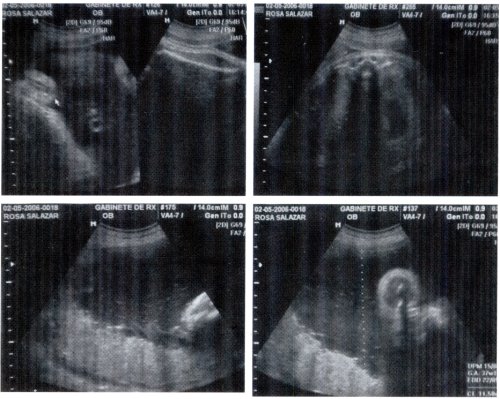
Fig. 2: geografía del 26 de abril de 2006: tiempo gestaeional de 36.5 semanas
3-05-06:
Ecografía obstétrica: Polihidramnios leve, placenta tópica, grado II. Depresión nasal de su raíz, prominencia de frente, compatible con S. de Apert ?.
Se solicita alfafetoproteina y antígeno carcinoembriona-rio. Ambos resultaron normales.
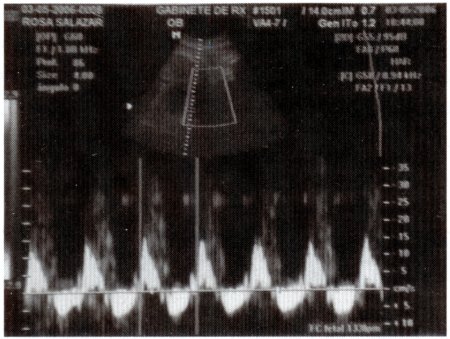
Fig. 3: Ecografía del 3 de mayo de 2006; tiempo gestacional de 38 semanas.
12-05-06:
Paciente que ingresa al servicio de emergencias referida de consulta externa, refiere dolor hipogástrico tipo contráctil. Al examen físico: AU 38 cm, se palpa producto en situación longitudinal dorso izquierdo, presentación cefálica. FCF: 148/pm; DU: aislada, cervix acortado, dilatado a 2 cm. presentación cefálica abocada al estrecho superior, bolsa íntegra. Se decide internación con diagnóstico de embarazo 38,2 semanas por U.P.M, fase latente de trabajo de parto, presenta pélvica, polihidramnios?
Se decide efectuar cesárea segmentaria con los diagnósticos mencionados, obteniéndose RN a término, de sexo femenino, peso 3280 grs., APGAR 8-9, liquido amniótico claro, aumentado de volumen, aproximadamente 1500 cc. placenta posterior fúndica. Al examen físico del RN, se constata: Cabeza con microcefalia y turricefalia, presencia de prominencia frontoparietal, fontanela bregmática de 5 cm, lamboidea de 2 cm; se aprecia deformación craneofacial; pabellones auriculares de implantación baja; tórax con retracción subcostal, extremidades superiores e inferiores con dedos cubiertos por membrana. (Sindactília en las cuatro extremidades).
Luego del nacimiento presenta quejido, con retracción subcostal, Silverman 2- 3. Se interna con diagnóstico de: RN AEG +- 40 semanas, Síndrome de distres respiratorio, taquípnea transitoria.
Conducta: Soluciones, oxigeno y exámenes complementarios:
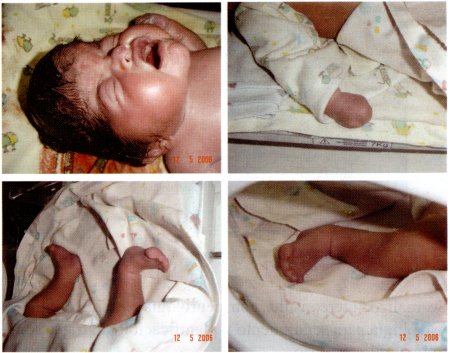
Fig. 4: Recien nacido el 12 de mayo de 2005.
TAC cerebral simple: se observa evidente hipotrofia de cerebro a nivel supratentorial, hay disminución de volumen de masa encefálica. Se aprecia agrandamiento de los ventrículos de cuerpo calloso. La sustancia blanca aparenta ser de mayor densidad.
Rx de cráneo: se observa displasia de estructuras craneales, el hueso occipital es aplanado y presenta como impresiones digitiformes.
Rx de manos y pies: En pies se aprecian a nivel del 2do metatarsiano aparente hueso supernumerario; En manos displasia ósea muy evidente, no se reconoce la anatomía ni la alineación de huesos.
Todos los signos clínicos hallados nos permitieron concluir que se trataba de una paciente con síndrome de Apert.

Fig. 5: Exámenes de gabinete
Se realizó interconsulta con neurología, traumatología y psicología para seguimiento y planificación del tratamiento integral necesario.
Después de 4 días de interacción la madre es dada de alta en buenas condiciones.
Al mes de vida se realizó la primera cirugía que consistió en: Avance de la barra fronto-orbitaria. Craneotomía de las suturas coronal y lambdoidea. Resección del pterion bilateral. Posteriormente se realizará corrección y plastia de manos y pies, avance facial a través de Lefort III.
COMENTARIO
El diagnóstico del síndrome de Apert se basa clásicamente en el hallazgo de una craneosinostosis primaria con cierre prematuro de las suturas coronal, sagital, escamosa y lamdoidea, acompañada de sindactilia de los dedos 2, 3 y 4, los cuales pueden estar fusionados al pulgar y al quinto dedo. Esta sindactilia puede aparecer también en los pies. Nuestra paciente presentó al examen físico sindactilia en miembros superiores "manos en cuchara", e inferiores, así como deformaciones craneofaciales.
El diagnóstico generalmente se lo realiza en edades tempranas, debido a que se trata de un trastorno del desarrollo. Este se fundamenta en criterios clínicos y radiológicos. Su detección temprana es importante, ya que el tratamiento por parte de los diferentes especialistas debe comenzar desde el momento del nacimiento, para obtener de esta manera resultados adecuados desde el punto de vista funcional, estético y psicosocial.
BIBLIOGRAFÍA
1. NELSON. Tratado de Pediatría, 1998, Vol.3, 15ava edición. Cap542.12 pag 2095. [ Links ]
2. David G. BROOKS, M.D., Ph.D.. Division of Medical Genetica University of Pennsylvania Medical Center. Philadelphia, PA. [ Links ]
3. Eugenio CARRO PUIG y Liliam S. FERNÁNDEZ BRAOJOS. Rev Cubana Pediatr 2005; 77(3-4) [ Links ]
4. Roger VOKAER. Traite d'Obstetrique. Pag 566 - 567. Ed. Masson, Paris. 1988 [ Links ]
5. BEISCHER, MACKAY, COLDITZ. Obstetricia y Neonatología. 3era edición. Graw-Hill Interamericana.Mexico 1997 [ Links ]
6. WILLIAM F. RAYBURN. JUSTIN P. LAVIN, JR. Obstetrics for the House Officer. Williams & Wilkins / Baltimore / London. 1994 [ Links ]

