











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkINTRODUCTION
Structural elucidation is an essential skill in organic chemistry and it is even more important in the field of natural products. The contribution of these studies are not only important to satisfy our scientific curiosity, it also has an invaluable contribution to drug discovery, since the natural products are often bioactive molecules and their structures are highly promising templates to design new medicines 1. Structural elucidation is also a very challenging field, it requires equal amounts of scientific knowledge, ingenuity and creativity. The chemist has to choose from hundreds of techniques and methods, to solve the intricate puzzle that is an unknown chemical structure. Nowadays, as a product of the technological advancements, the organic chemists have powerful spectroscopic tools to achieve such a goal, which turns this task into routine procedure in every lab around the world. But just six decades ago, it was a titanic task, because the instruments needed to measure the spectrometric data were not available or still under development. Thereby, a primary method in the structural elucidation of natural products was the use of degradative reactions using the pure natural substance as starting point, as well as isotopic labeling and sometimes synthetic reactions. In the present article, we have taken advantage of old publication 2 to discuss some organic reactions from the mechanistic view point, this time, to explain its use to elucidate the partial structure of a complex natural product, Withaferin A. Due to lack of knowledge of classical mechanisms, students experiment problems to comprehend a determined synthesis or chemical reaction. Since a mechanistic proposal is naturally mandatory for a rational explanation of products emerging from an organic reaction, we offer the present series theming on mechanistic approaches on several published syntheses. As academics we are committed with the didactics and we have designed a series of articles exposing mechanistic theoretical proposals, where articles have a character of analytical review: The Organic Chemistry Notebook Series, a Didactical Approach.
DISCUSSION
LITERATURE REVIEW AND THE CORRESPONDING MECHANISTIC THEORETICAL APPROACH
Isolation, derivatization, Infrared (IR), Ultraviolet (UV)
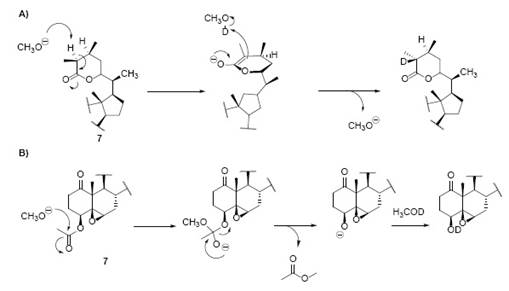
Withania somnifera Dun. has been extensively studied regarding its chemical constituents, mostly by Israeli scientists during the sixties and earlies 1970 decade in the series of structural studies denominated: “Constituents of Withania somnifera Dun.”. The goal of such study was the discovery and structural elucidation of withaferin A, established in three papers discussing the side chain [2], the planar structure 3, and the stereochemistry 4 of the compound. Hence, we will present a theoretical analysis of the structural elucidation of withaferin A, starting from the determination of the structure of the side chain 2. This study 2) seems to be the first approach to the structural determination of withaferin A, not only because of its year of publication (circa 1965), but for the analysis developed therein.
Various natural substances were isolated from the leaves of W. somnifera, mainly two withanolides: withaferin A 1 (A 2 1) and dihydrowithaferin A 2 (A 3 1) [2]. For a description of the reaction pathways see Fig. 1. 1 was first isolated as white crystals, λmax = 214 mμ, (( 17,500), and vmax = 1692 cm-1 (2 . Elemental analysis afforded a molecular formula of C28H38O62. The chemical, elemental analysis and spectroscopic assays on 1, included acetylation and the stepwise hydrogenation with three moles of hydrogen over palladium as catalyst. The acetylation afforded withaferin A diacetate 3 ([A 2 - 2OH + 2OAc], see Fig. 1 and 2) corresponding to two acetylable hydroxyl groups, one of them being primary as demonstrated later by the hydrogenolysis that yielded 62.
The first hydrogenation on 1 added one mol of H2 yielding dihydrowithaferin A (2) as a reduced derivative of 1, which according to their 1H-NMR spectra, resulted identical to 2 (original sample). The 2H difference between 1 and 2 was confirmed by IR, due the hypsochromic shift from λmax = 214 to 210 mμ. Additionally, the IR, showed absorption peaks at 1692 cm-1 (1) and 1700 cm-1 (2) respectively, indicating the presence of two different carbonyl groups (α,β -unsaturated ketone and δ-lactone) in 1, and only one peak in 2 corresponding to both carbonyl groups 2. The second hydrogenation on 2 produced the hydrogenolysis of the primary alcohol, corresponding to the reduction of the hydroxyl group in the allylic position yielding dihydrodesoxywithaferin A 4 [A 3 + H + H2O]. This hydroxyl group loss was confirmed by the formation of the dihydrodesoxywithaferin A monoacetate 6 ([A 3 + H + H2O - OH + OAc]) by direct acetylation. On another pathway, the acetylation on 2 produced dihydrowithaferin A diacetate 5 ([A 3 - 2OH + 2OAc]) which after a second hydrogenation, gave the hydrogenolysis of the methyleneacetyl group into a methyl group and acetic acid to yield dihydrodesoxywithaferin A monoacetate 6 ([A 3 - 2OH + OAc + H + HOAc]). Both products, [A 3 - OH + OAc + H + H2O] and [A 3 - 2OH + OAc + H + HOAc] proved to be the same product 6 by TLC analysis and 1H-NMR. After a third hydrogenation on 6, yielded tetrahydrodesoxywithaferin A monoacetate 7, symbolized by [A 3 + H + H2O - OH + OAc + H2] and [A 3 - 2OH + OAc + H + HOAc + H2], respectively. Both products proved once more being the same product 7, due to lacking of any strong UV absorption, and the presence of two IR bands in the carbonyl region at 1739 and 1715 cm-1. All the data indicated the presence of two C=C double bonds which are part of the α,β-unsaturated ketones in 1.
Nuclear Magnetic Resonance (1H-NMR)
The up-field region of the 1H-NMR spectrum of 1 [2], showed three singlets corresponding to three tertiary methyl groups (δH = 0.68 ppm, 3H; 1.38 ppm, 3H; 2.03 ppm, 3H), and one doublet, for a secondary vinylic methyl group (δH 0.97, d, 6.5 Hz, 3H). A signal for an hidroxymethylene group (δH 4.35, s, 2H), this primary alcohol has a shift toward downfield until δH 4.87 in the diacetylated derivative 3. The acetylation of this alcohol positions was carried out with acetic anhydride, the mechanism is shown in Fig. 2. At downfield, two clearly distinguishable signals appear as a double doublet (δH = 6.97 ppm, 10, 6 Hz, 1H) and a doublet (δH = 6.18 ppm, 10 Hz, 1H), corresponding to two protons in a cis-doble bond. At upper field, a proton (δH = 3.75 ppm, d, 6 Hz, 1H) is coupled to a proton at δH = 6.97 ppm (J = 6 Hz). The chemical shift of this proton (δH 3.75) suggest that is geminal to an oxygen, appearing shifted at δH 4.66 after acetylation, confirming a secondary alcohol function. The doublet multiplicity of its signal means that the proton is no further coupled to any other proton. This three-proton coupling system corresponds to cycle A of withanolides 5. The first hydrogenation applied on 1 gave 2 (A 3 ), (Fig. 1) was confirmed by the disappearance of these downfield signals in 2 (δH = 6.97 and 6.18 ppm). The hydrogenation mechanism is the usual for the catalytic (Pd) hydrogenation of a double bond, as shown in Fig. 3. The signal of H-4 changed from a doublet into a triplet and shifted to higher field at δH = 3.55 ppm. In the corresponding diacetate 5, this proton has suffered a downfield shift from δ = 3.55 to 4.62 ppm appearing now as triplet 2.
The second hydrogenation (Fig. 1) or the hydrogenolysis of the -CH2OH (C-27) group was confirmed by the disappearance of the signal at δH = 4.35 ppm geminal to an -OH group and the appearance of a new singlet corresponding to a vinylic methyl group at δH = 1.93 ppm. The proposed mechanism for this reaction is shown in Fig. 4 6. The authors mentioned regarding the composition of the side chain of 1 the following after the third hydrogenation:
“In tetrahydrodesoxywithaferin A monoacetate 72, these two methyl groups are shifted up-field and appear now as two new sets of doublets centered at δH = 0.92 and 1.13 ppm indicating unequivocally that the two vinylic methyl groups in dihydrodesoxywithaferin A 6 2 are both located on the same double bond, and therefore, in withaferin A 1 2, the primary alcohol and the vinylic methyl group are placed as well on the same double bond 2. These conclusions imply that from the molecular formula previously established (C28H38O6), two oxygen atoms have proven to belong to the two alcohol groups, two other are in the carbonyl functions already established. The two remaining oxygen atoms are cyclic ethers, one probably in a lactone moiety, due to the existence of a carbonyl group and an epoxide whose presence was determined by chemical means in a later publication 7.
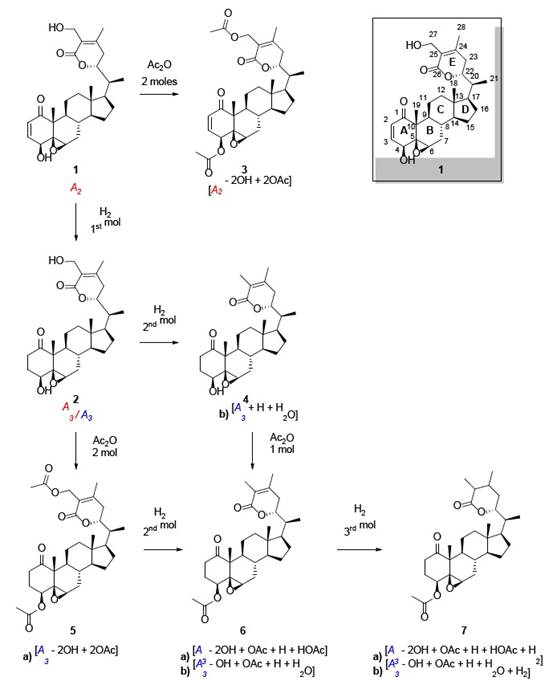
Fig. 1 Withaferin A (1) and dihydrowithaferin A (2): Description of the stepwise hydrogenations and several acetylations [2].
There is an alternative interpretation in which two oxygen atoms belong to secondary alcohol functions, already demonstrated, one belongs to a carbonyl in cycle A, the fourth is the epoxide geminal to the unchanged proton (δ 3.20) throughout the different derivatives. The two last oxygen atoms belong to a δ-lactone ring or side chain; whose demonstration follows below 2. The establishment of the β stereochemistry of the C5-O-C6 group was published later 4.
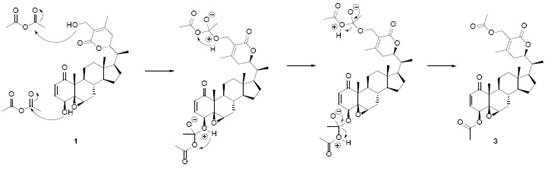
Fig. 2 Reaction mechanism of the acetylation of withaferin A (1), yielding the diacetyl derivative, withaferin A diacetate (3).
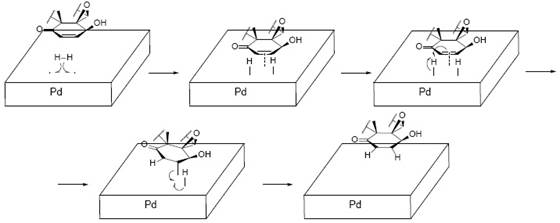
Fig. 3 Withaferin A (1), mechanism of the hydrogenation (1st) of the α,β-unsaturated ketone (Ring A), to yield derivative dihydrowithaferin A, (2).
The side chain of withaferin A (1), dihydrowithaferin A (2) and dihydrodesoxywithaferin A (4)
For all three compounds (1, 2, 4) (See Fig 1 and 4), H-22 was assigned the value δ = 4.40 ppm and the multiplicity dt2. Despite the fact that the maximal UV absorption λ max = 210 nm in 2 is not compatible with such structure (2), the value λmax = 226 nm in 4 is appropriate, for an α,β-unsaturated lactone 2. The hypsochromic shift of 16 nm in 2 with respect to 4 correspond to the primary alcohol at C2-CH2OH 2.
Hydrolysis of the lactone ring
The attempts to open the δ-lactone in 4 to form the corresponding hydroxycarboxylic compound by hydrolysis, failed 2. Reactions included alkaline and acidic hydrolysis. If ring-opening took place, it was not possible to avoid its reversibility 2.
Reduction of tetrahydrodesoxywithaferin A acetate (7)
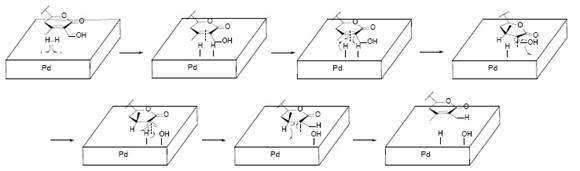
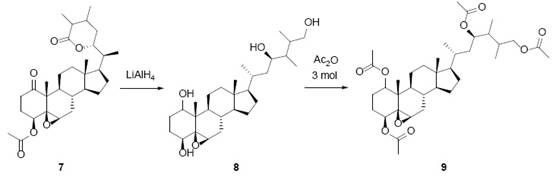
Tetrahydrodesoxywitaferin A acetate (9, Fig. 5) was submitted to reduction with LiAlH4 (Fig. 5). The product of the reduction was a tetrol (8, Fig. 5) where two secondary alcohol groups were formed from the acetate and carbonyl group 2. The other two alcohol groups, a primary and a secondary alcohol, were product of the reductive opening of the lactone 2. The mechanism is the usual for reduction of ketones and esters, as shown in Fig. 6. The 1H-NMR spectrum of 9, exhibited three one-proton singlets: δ = 4.48. 4.88 and 5.00 ppm. The last one (δ=5.00 ppm) was previously a double triplet being assigned to H-22 in 8 (δ 4.22 ppm), therefore it shifted to downfield after acetylation of 8. The remaining protons in 9 (δ 4.48 ppm and 4.88 ppm) were not assigned 2.
Deuterium exchange experiments, deuteration at C-24
The 1H-NMR for H-22, shows that this proton is part of an ABX system (Fig. 7), where H-22 is a double triplet [2]. There two different interpretations of this system, isomers 4 and 10 (Fig. 7). In case of 10 we have only one allylic proton involved instead of two allylic protons as it’s the case in 4 [2].
In order to confirm one of this isomeric forms, deuterium-exchange experiments were undertaken [2]. Dihydrodesoxywithaferin A (4) was submitted to a deuterium-exchange reaction by using deuterated methanol and sodium methoxide. The product contained at least five deuterium atoms after replacement of 5 hydrogens, as observed by the mass spectrometry 2. The 1H-NMR of the deuterated compound 4a shows now a doublet for H-22 (δ 4.37, J 4Hz), meaning that this proton is coupling with just one neighbor. The authors of [2] deduce from this, that the two allylic protons on C-23 were exchanged by deuterium, discarding isomer 10a as a possible structure (Fig. 7). The other three deuterium units occupy one of the vinylic methyl groups 2.
In order, to be confirm that the deuteration took place at the allylic position in 4a, (Fig. 7), tetrahydrodesoxywithaferin A (7) acetate, where no double C=C bonds are present, was treated under the same deuterium-exchange reaction [16]. The reaction did not exchange deuterium in the lactonic ring, which was evident since after re-acetylation, proton H-22 had the same multiplicity (dt) as in 7 [16].
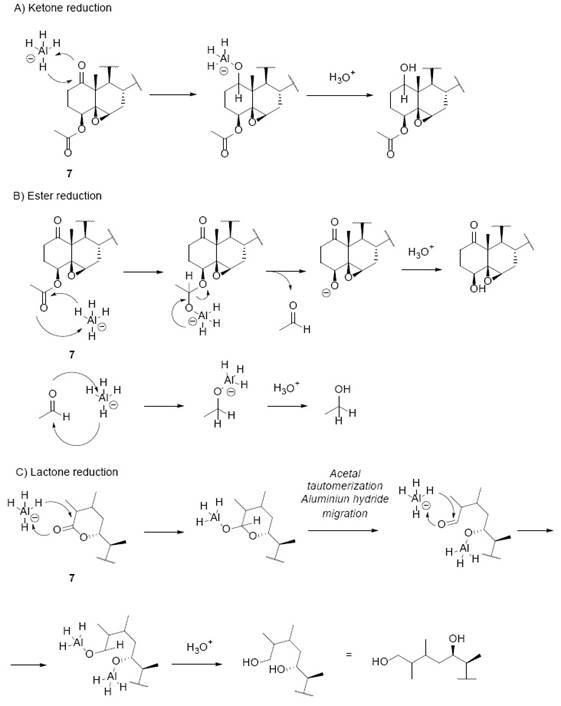
Fig. 6 The reduction process of tetrahydrodesoxywithaferin A acetate (7). A) Reduction mechanism of a ketone by LiAlH4. B) Reduction mechanism of an acetate (ester)by LiAlH4. C) Reduction mechanism of a lactone by LiAlH4.
Nonetheless, the fact that the coupling of the C-23 allylic methylene protons and H-22 was previously established by the authors by decoupling experiments on Withaferin A diacetate (3) [2]. The 1H-NMR spectrym of 3 showed a dt at 4.41 ppm (J = 12. Hz) which was correlated to two multiplets (undefined splitting patterns) separated by 12.5 Hz and centered at 2.37 ppm [2]. Irradiation at 2.37 ppm provoked the collapse of the double triplet at 4.41 ppm into a doublet [2]. The chemical shift of 2.37 ppm falls in the range for allylic protons 8, confirming that H-22 (δ = 4.41 ppm) is coupling with them. The deuterium exchange mechanism for the allylic positions is shown in Fig. 8.
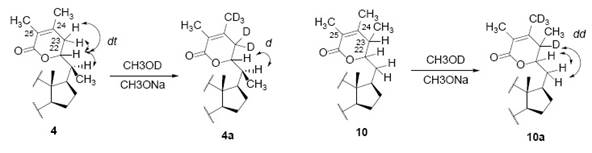
Fig. 7 Possible isomers for the side chain in dihydrodesoxywithaferin A (4) and thereby in withaferin A (1). The expected results after deuterium exchange are shown as 4a and 10a. The 1H-NMR multiplicities for H-22 are shown next to the proton.
Before deuteration, the stability to alkaline medium of 7 was tested [2]. This treatment was done with potassium hydroxide; therefore the reactive groups will be 4-OAc (ring A) and the lactonic ring, as shown in Fig. 9. The alkali action on the acetyl function was hydrolized to a secondary alcohol (See Fig. 2) and the lactonic ring would eventually yield a hydroxy-acid (Fig. 9). The acid treatment (pH 3) of this product, would regenerate the lactonic ring, in a deacetylated product of 7a (Fig. 9). It can be deduced from Fig. 9 and following the reaction mechanism, that the product obtained after alkalization is a deacetyl hydroxyl-acid derivative of 7. After opening of the ring, a regeneration of the lactone was observed under pH = 3. Subsequent acetylation of 7a yielded a product identical to 7. This is the same kind of result to that reported for the hydrolysis of the lactone of 4, as described in the experimental section 2. Then authors propose that an isomeric derivative was obtained, after alkalization of 7, but there is not experimental evidence that confirm it, and it could be the starting product 72. Neither the main text nor the experimental section of 2, show any details of about the characterization of the isomeric product of 7.
The incorporation of 5 deuterium atoms in the side chain was confirmed by MS spectrometry after deuteration of 42. Three deuterium atoms were exchanged at one of the vinylic methyl groups and the other two deuterium were assigned to the allylic protons at C-23 in 4 proven by the fact described as: “the deuterated compound indicates unequivocally in its n.m.r. spectrum that the C-223 proton displays now a doublet at δ 4.37 (J = 4 c.p.s) and has therefore only one neighboring proton; two allylic hydrogens which were previously present in the molecule have then been exchanged with deuterium and the part structure IV could be eliminated” 2.
Epimerization of tetrahydrodesoxywithaferin A acetate (7)
The alkaline treatment of lactones can provoke epimerization in a carbon next to a carbonyl function [9], C-25 of the lactone ring [2]. The epimerization takes place through an intermediate enolate, that rearranges to the most stable epimer 2. It has been proposed by the authors [2] that hydrogens H-25 and H-26 in the lactone should be positioned in a cis isomerism, after the catalytic hydrogenation of the C24=C25 double bond in 6, as shown in Fig. 10 by a syn hydrogen addtion. Therefore, the isomerism of H-25 and H-26 should be in a trans in the epimer 2. The epimerization can be observed by 1H-NMR, the signal on the secondary methyl group C-27 shifted from δ = 0.92 (d, J = 6.5 Hz) in 7 to δ 1.32 (d, J 5.7 Hz) in 7b [2]. The formed epimer 7a, can form enolates between C-25 and C-26 after treatment with a base like sodium methoxide 2.Therefore, when the enolate can react with deuterated methanol, the deuterium is incorporated on C-25, because of that the methyl C-27 is now observed now as a singlet 2.
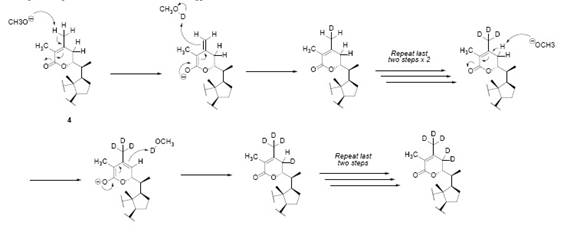

Fig. 11 shows the mechanism for the inversion of methyl C-27. This configuration change is visible by the chemical shift of this methyl group toward downfield mentioned in the article under current examination (δ = 0.92 - 1.32). The authors of [16] do not mention this observation. They do not propose any assignment to the signals of 1H-NMR regarding the methyl groups, neither before nor after epimerization. The mechanism (Fig. 11) goes through the reaction of the acidic H-25 with the base forms the carbanion that provokes the enol-carbonyl equilibrium, in the last step a proton is added to the enol. It is important to observe that the anti-addition is favored due the steric hindrance between both methyl groups, ending with the trans isomer.
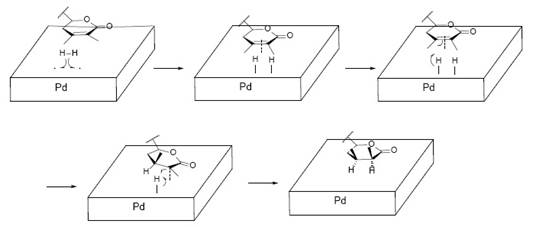
Fig. 10 Catalytic hydrogenation from 6 to 7, the syn hydrogen addition, form a cis configuration in the product.

Deuterium exchange experiments, deuteration at C-2 (C2-H) in the side chain of tetrahydrodesoxywithaferin A acetate (7) during epimerization
The deuteration process, has so far been explained for two different positions inside of the side chain, CH2-24 (4) and CH3-27 (4). In this section we will explain the deuterium exchange of C-25 of 7, during the epimerization ( cf. Epimerization of tetrahydrodesoxywithaferin A acetate (7) ) [2], the epimerization mechanism was proposed on Fig. 11.
As well as 4, compound 7 was treated with deuterated methanol, for deuterium exchange in 7. The apparition of a temporary enolate is involved (See Fig.12, A) and consequently, the exchange of the C-25 hydrogen by deuterium (Fig. 12, A). This is observed in the 1H-NMR of the deuterated epimer, where the singlet originally (before isotopic exchange) at δ = 0.92 ppm was shifted downfield to δ = 1.30 ppm 2. A consequence of the treatment with sodium ethoxide, is the deacetylation (Fig 12, B) on C4-OH. The re-acetylation process goes in the same way than the described on Fig. 2.
Five-, seven- or six-membered α,β-unsaturated lactone ring?
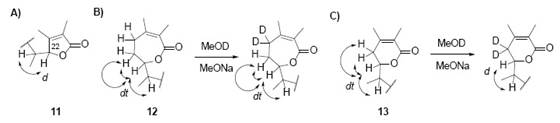
It seems legitimate to make such question, fortunately, threre are experimental results to find an answer 2. Fig. 13 shows structures 11, 12 and 13 (five-, seven- and six-membered lactone rings). “In former (114) the corresponding C-5 proton could not possibly display a double triplet while in the latter (12 4 ) the corresponding double triplet should have remained unchanged following the deuteration experiments” [2]. The evidence for the option of the six- membered α,β-unsaturated lactone ring derives from data of 1H-NMR. Isomer 11 finished being rejected because, without deuteration, the 1H-NMR should show a doublet multiplicity for H-22, but the experimental signal is a doublet triplet. 12 was rejected because after deuteration (see Fig. X), it should show a double triplet multiplicity, but experimentally is a doublet. 13 complies with all 1H-NMR data, therefore is the correct structure.
The IR spectra of tetrahydrodesoxywithaferin A acetate (7)
IR spectra show absorption bands at 1739 cm-1 that corresponds to one acetate and one lactone groups, and 1715 cm-1 that corresponds to two carbonyl groups See Fig. 14. After epimerization (see Fig.11) and deacetylation (see Fig. 12) the absorption bands were 1712 cm-1 corresponding to just two carbonyl groups (See Fig. 14), one ketone and one lactone groups. The overlapping bands at 1712 cm-1 were resolved in two bands at 1718 and 1706 cm-1, through the use of a KBr pellet, (ketone and lactone irrespectively). After re-acetylation (See Fig. 2) the bands are at 1730 cm-1 (acetate) and 1712 cm-1 (overlapping of ketone and lactone) 2.
Ozonolysis of dihydrodesoxywithaferin A acetate (6)
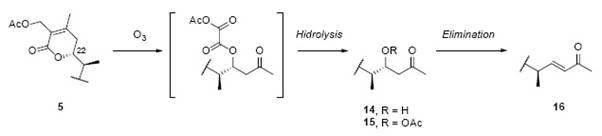
Another reaction to demonstrate the structure of the α,β-unsaturated lactone was the ozonolysis of 6, see Fig. 15 2. The reductive decomposition of the intermediate ozonide yielded the β-hydroxy ketone 14, characterized as its crystalline acetilated derivative 152. Neither, 14 nor 15 showed UV absorption, proving the disappearance of the chromophopre group 2. The 1H-NMR showed the disappearance of the vinylic methyl groups and the presence of a new α-methyl ketone group [16].

Fig. 14 The IR bands for the carbonyl groups in tetrahydrodesoxywithaferin A acetate (7) and its epimer (7b)
The signal of H-22 shifted from δ = 4.41 ppm in 6 to δ = 4.11 ppm in 14, and a shift to δ = 5.36 ppm in 15, all signals exhibited a double triplet multiplicity [16]. The next step in the degradation pathway was the dehydration of 14 into the α,β-unsaturated methyl ketone 16. Three 1H-NMR spectra, shows a H-23 signal at δ = 5.98 ppm (d, J ab = 16 Hz, Ha), and a H-23 signal at δ = 6.67 (q, J ba = 16 Hz, J bc = 8 Hz, Hb). The coupling constant value of 16 Hz, confirms a trans alkenic system [2].
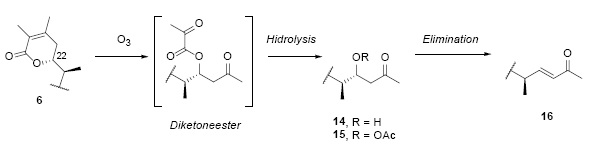
The mechanism in Fig. 16 is based on the ozonolysis mechanisms published by Criegee et al [10] and Geletneky et al [11]. The experimental section ( cf. Ozonolysis of Dihydrodesoxywithaferin A Acetate (6) ) explains that 6 was dissolved in a mixture of chloroform and ethyl acetate [2]. This implies a liquid medium for the reactions; hence mechanism excludes a free radical process in favor of an ionic one. Ozone O3 was then passed through the solution at 0°C. As shown in Fig. 16, two oxygen atoms of ozone were added to the C=C double bond in the lactone ring to yield an adduct comprising a five membered heterocycle, called molozonide. This kind of heterocycles is unstable intermediates and will further undergo to form the ozonide bicycle. After concentration the crude ozonide is dissolved in aqueous acetic acid and reduced by powdered metal zinc and forms a precipitate of zinc oxide separated by filtration. At this point the aqueous media contains the intermediate diketoneester and acetic acid, which leads to the hydrolysis of the ester and releasing pyruvic acid and giving 14 as product.
Preparation of the α,β-Unsaturated Ketone (16)
The continuation of this derivatization was done by dehydration of 14 as mentioned in cf. Preparation of the α,β- Unsaturated Ketone VII2. The hydroxyketone 14 was diluted in chloroform/benzene and contained in a column with acid-treated (butanedioic acid) alumina for 3 days 2. The product was then washed out with chloroform to yield (16). This compound showed IR signals at 1733 cm-1 (acetate), 1718 cm-1 (ketone), 1672 cm-1 and 1642 cm-1 (unsaturated methyl ketone). Fig. 28 shows the reaction mechanism and the treatment of alumina with butanedioic acid. These conditions generate a medium slightly acid (pH 6.7) 12.

Fig. 16 Ozonolysis of dihydrodesoxywithaferin A acetate (6), degradation of the side chain of 6 into 14.
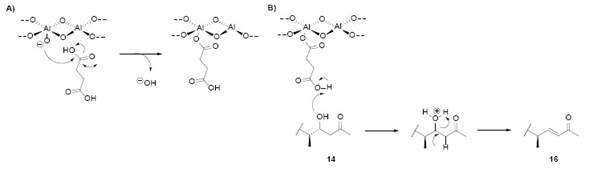
Fig. 17. A) Acidification of alumina by butanedioic acid; B) Deshydratation mechanism of 14 by the acid-treated alumina.
Position of the primary alcohol group on the lactone ring of Withaferin A (1), either at C-27 or at C-28
“There remained to be determined the position of the primary alcohol group on the lactone ring of Withaferin A, either at C-275 or at C-285 (Cf. Ia)” [16]. Before determining the position of the primary alcohol in Withaferin A, we mention that dihydrowithaferin A diacetate (5) was obtained from dihydrowithaferin A (2). The diacetate comes from acetylation of the two alcohol functions in 2. This synthesis was useful for proving the structure of the side chain by ozonolysis, as described in the next section.
Ozonolysis of dihydrowithaferin A diacetate (5)
Compound 5 was submitted to ozonolysis following the same procedure than 6 [cf. Ozonolysis of dihydrodesoxywithaferin A acetate (6) ] 2. The product obtained was similar to that from the ozonolysis of 6, see Fig.18 [16]. The mechanism is the same as for 6 (Fig. 16).
Preparation of the α,β-Unsaturated Ketone VII from 5
The product 14 was obtained from the ozonolysis of 5 (See Fig. 18) and it was identical to the obtained from 6 [2]. These results allow establishing the position of the primary alcohol function on C-27 of the δ-lactone. The ozonolysis of 5 and 6 gave both the same product 14 and same derivatives 15 and 16. These three were identified structurally by spectroscopic methods, as a hydroxymethyl ketone (14), hydroxymethyl ketone acetylated (15) and a α,β-unsaturated methyl ketone (16) (Fig. 15). These products are all methyl ketones and not hydroxy- or acetoxy- methylene ketones indicates that C-28 has a methyl group as substituent. The authors in 2 conclude “Should the alcohol group have been placed in the alternative (to C-275) C-285 position ozonolytic cleavage would result in an α- acetoxy ketone, RCH(CH3)CH(OH)CH2COCH2OAc” 2.
A supplementary treatment of cleavage of the double bond, the osmium tetroxide-sodium periodate procedure.
This reaction was tested on compound 6 [2]. The same product 16 was obtained, confirming C-27 as the position for the primary alcohol function 2. Fig. 19 shows the mechanisms involved in the formation of the diketone ester (Fig. 16) and acetyldiketone ester (Fig. 18) after the oxidation of 5 and 6 with OsO4, was followed by the oxidative cleave of the diol by NaIO4 (See Fig. 19).
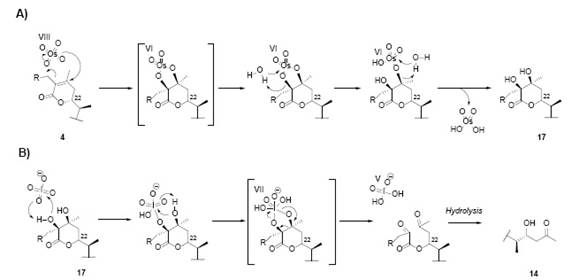
Fig. 19 Cleavage of the double bond by the osmium tetroxide and sodium periodate procedure, dihydrodesoxywithaferin A acetate (5) and dihydrowithaferin A diacetate (6), A) dioll formation mechanism by OsO4 and B) oxidative cleavage mechanism of the diol by NaIO4, followed by the ester hydrolysis to yield 14.
Electronic Impact Mass Spectrometry analysis
EIMS assays were performed on Withaferin A (1), dihydridesoxywithaferin A (4) and dihydridesoxywithaferin A monoacetate (6), the results confirmed the structural elucidation discussed previously 2 and analyzed in the present paper.
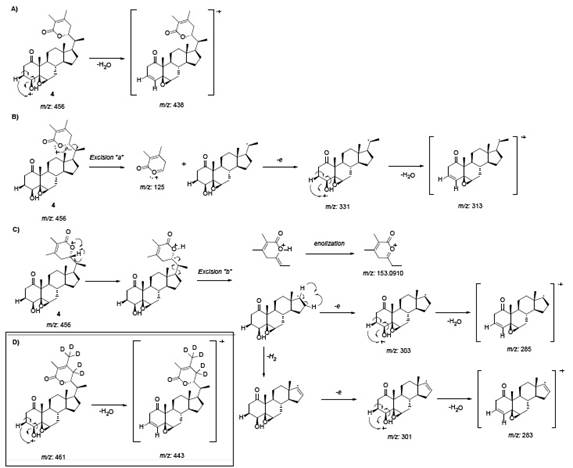
Compound 4 showed among the next relevant MS peaks: m/z 456, 438, 331, 313, 303 and 285. The highest mass peak m/z 456 shows a molecular formula of C28H40O5, coincident with the previous results from elemental analysis 2. The peak m/z 438 is obtained by eliminating water from the secondary alcohol function on C-4, the mechanism is shown in Fig. 20A. Excision “a” (See Fig. 20B) forms fragments m/z 331 and m/z 313 due to cleavage of the lactonic ring and followed by the loss of water, respectively [2]. Excision “b” (See Fig. 20C) forms fragments m/z 303 and m/z 285 corresponding to the loss of the whole side chain and followed loss of water, respectively, this mechanism was inspired by previous MS studies on withanolides 13. Loss of H2 in the m/z 303 fragment, yields the fragment at m/z 301, which after losing water gives the fragment m/z 283. “In the deuterated dihydrodesoxywithaferin A loss of elements of water gave rise to the peak m/z 443 indicating an exchange introducing five deuterium atoms. All other peaks were identical with the corresponding ones in the nondeuterated compound, indicating thereby un equivocally that deuterium exchange involved the lactone ring only” 2. The proposed mechanism for the deuterated derivative is shown in Fig. 20D.
CONCLUSION
This review has analytical character, we mean by that that besides the bibliographical review supporting it, it provides commentaries and new apports on the synthesis reported by authors of the reviewed articles, from the theoretical mechanistic point of view. It also opens a window to a historical primary methodology of natural products research with a deepening in organic reactions of current use with such purpose at the time.

