










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO  uBio
uBio Share

 Permalink
PermalinkRevista Boliviana de Química
On-line version ISSN 0250-5460
Rev. Bol. Quim vol.37 no.1 La Paz Apr. 2020
https://doi.org/10.34098/2078-3949.37.1.7
FULL ORIGINAL ARTICLES
Hydroboration, a brief historical review through mechanistic views, part I:
alkyl-and aryl-substituted olefins, as addition-substrates; the organic chemistry notebook, N ° 15
Hidroboración, una breve revisión histórica a través de vistas mecanicistas,
parte I: olefinas alifáticas y aromáticas como sustratos de adición; el cuaderno de
química orgánica, N ° 15
José A. Bravo1'*, José L. Vila2
1Natural Product Laboratory, Phytochemistry, Chemical Sciences Department, Instituto de Investigaciones Químicas IIQ,
Facultad de Ciencias Puras y Naturales FCPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303,
Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, phone +59122792238,
La Paz, Bolivia, jabravo@umsa.bo, joseabravo@outlook.com, www.umsa.bo
*Correspond¡ng author: iabravo@umsa.bo, ioseabravo@outlook.com
2Natural Product Laboratory, Synthesis and Hemisynthesis, Chemical Sciences Department, Instituto de Investigaciones Químicas IIQ,
Facultad de Ciencias Puras y Naturales FCPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303,
Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, phone +59122772269,
La Paz, Bolivia, jlvila@umsa.bo, joselu62@hotmail.com, www.umsa.bo
Received 09 11 2019 Accepted 04 19 2020 Published 04 30 2020
Abstract
The Organic Chemistry Notebook Series, a Didactical Approach, is the series designed with educational purposes in the organic synthesis field. With the present paper we add to a total of fourteen contributions so far in the series.
This series of studies is designed to help students when getting started in the synthesis subject. The method of learning includes many fully and explicitly designed reactions step by step. The best manner to understand a synthesis is by means of graphical views, in this case, the ones proposed by the authors of this series, and, when in most of the cases they are accompanied by illustrative comments that describe the graphical mechanistic proposals and add some criteria deduced from the different mechanistic steps. During the past 14 chapters we have taken a series of reactions compiled by W. Carruthers in 'Some modern methods of organic synthesis', and we have proposed didactical and mechanistic views for them. Now we are boarding another important compiling source in the svnthesis studies: 'Advanced Oreanic Chemistrv. Part B: Reaction and Svnthesis' bv Francis A. Carev and Richard J. Sundberg. This theme is included in the chapter "Electrophilic additions to carbon-carbon múltiple bonds", and references therein.
Keywords: Organic Chemistry, Addition reaction, Múltiple bonds, Organoboranes, Hydroboration, Mechanisms of Reactions.
Resumen
La Serie de Cuadernos de Química Orgánica, un Enfoque Didáctico, es la serie diseñada con fines educativos en el campo de la síntesis orgánica. Con el presente trabajo agregamos un total de quince contribuciones hasta ahora en la serie.
Esta serie de estudios está diseñada para ayudar a los estudiantes cuando se inician en el tema de síntesis. El método de aprendizaje incluye muchas reacciones diseñadas de forma completa y explícita paso a paso. La mejor manera de comprender una síntesis es mediante vistas gráficas, en este caso, las propuestas por los autores de esta serie y, cuando en la mayoría de los casos, van acompañadas de comentarios ilustrativos que describen las propuestas gráficas mecanicistas y agregan algunos criterios deducidos de los diferentes pasos mecanicistas. Durante los 14 capítulos anteriores, hemos tomado una serie de reacciones compiladas por W. Carruthers en "Algunos métodos modernos de síntesis orgánica", y hemos propuesto puntos de vista didácticos y mecanicistas. Ahora abordamos otra fuente importante en los estudios de síntesis: "Química orgánica avanzada, Parte B: Reacción y síntesis" de Francis A. Carey y Richard J. Sundberg". Este tema se incluye en el capítulo "Adiciones electrofílicas a los enlaces múltiples carbono-carbono" en el texto mencionado y referencias incluidas.
Palabras clave: Química orgánica, Reacción de adición, Enlaces múltiples, Organoboranos, Hidroboración, Mecanismos de reacción.
INTRODUCTION
Due to lack of knowledge of classical mechanisms, students experiment emptiness when comprehending a determined synthesis found in the literature. Since a mechanistic proposal is naturally mandatory for a rational explanation of producís emerging from a synthesis, we offer the present series theming on mechanistic approaches on several published syntheses. As academics we are committed with the didactics and we have designed a series of articles exposing mechanistic theoretical proposals, arricies have a character of short review. The present contribution: Hydroboration, a brief historical review through mechanistic views, part I: alkyl- and aryl-substituted olefins, as addition-substrates, The Organic Chemistry Notebook, N° 15, that refers to regioselectivity considerations, is the fifteenth study in the series: "The Organic Chemistry Notebook Series, a Didactical Approach" [1-14].
REVIEW OF REACTIONS, MECHANISTIC THEORETICAL PROPOSALS, DISCUSSION
Hydroboration of olefins
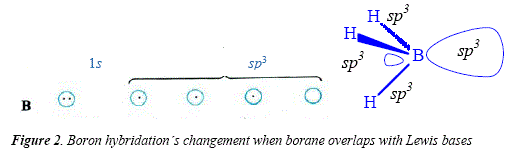
Hydroboration is a good method in many synthesis reactions employing electron-rich compounds as substrate, namely alkenes and alkynes [15]. Boron as borane BH3 is a Lewis acid with a free orbital suitable for rooming an electrón pair. Borane is available as a dimer, B2H6, with the boron atoms bridged by two of the six hydrogens between them [15]. Due to its Lewis acid nature (Figure 1), boron interaets with Lewis bases such as oxygen, nitrogen or sulphur in ethers, tertiary amines and sulfides, R2O+--BH3, R3N+--BH3, R2S+--BH3 [15], see boron hybridation's changement when borane overlaps with Lewis bases (Figure 2).
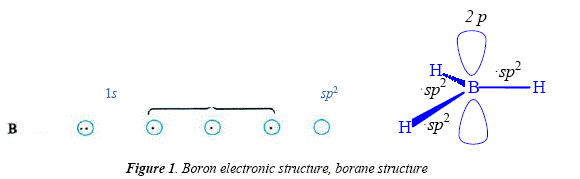
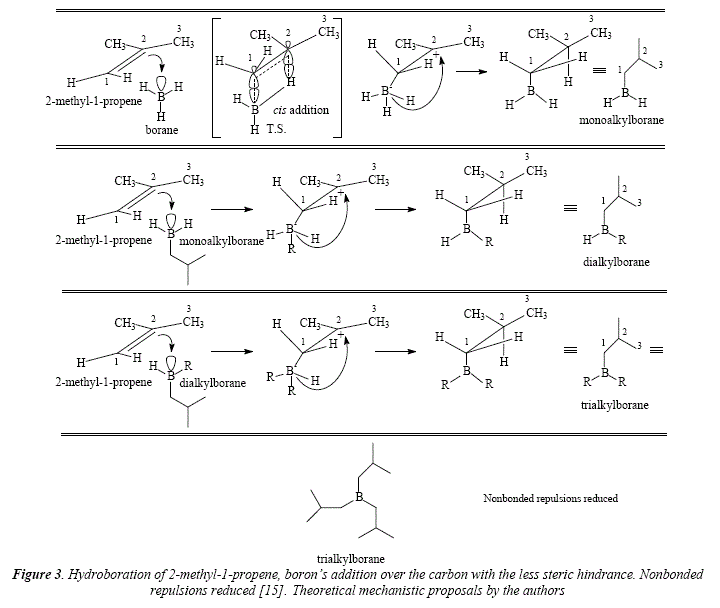
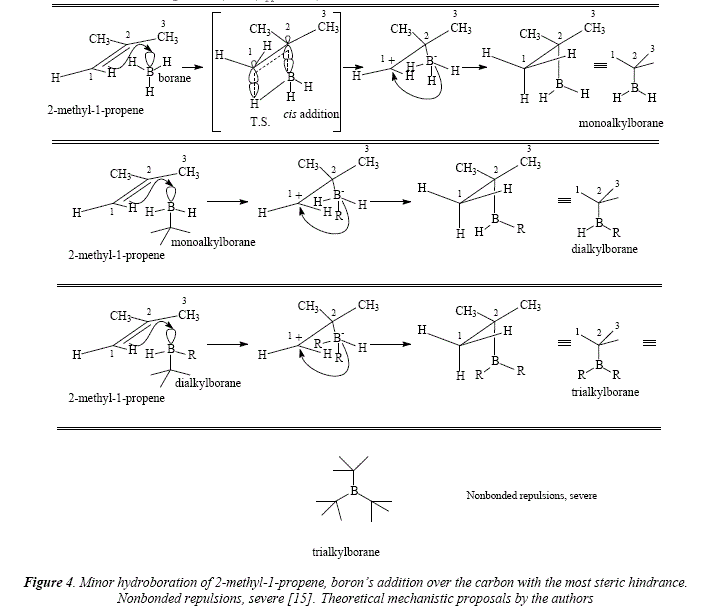
Using THF as solvent or DMS, the addition of borane over most alkenes is immediate [15], a reaction known as hydroboration. Hydroboration is stereospecific and very regioselective [15]. Steric and electronic effects are involved in the addition of borane and they favor the attack over the carbón the less substituted [15]. Since borane reacts with alkenes until completion of the hydride substitution in BH3 and their replacement by the adding on three alkene molecules to give a trialkyl borane, the less substituted carbón is chosen by boron in order to avoid steric hindrance when adding the second and the third alkenic residue, e.g. the addition of BH3 to 2-methyl-1-propene, where atom 1 is preferred over atom 2 for the new B-C bond [15], see Figure 3. This behavior gives this reaction its regioselective quality [15]. The minor option of addition of the boron atom to the carbón with the most steric hindrance represents severe nonbonded repulsions, see Figure 4.
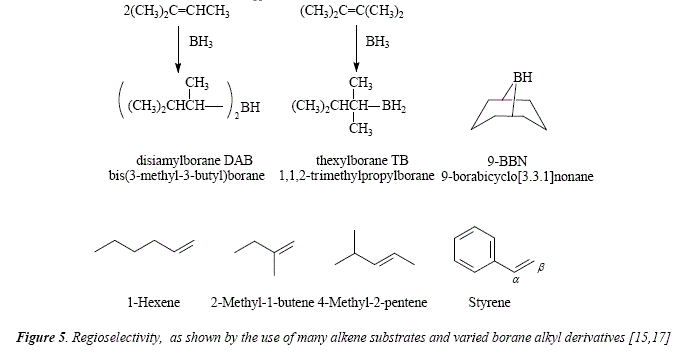
The regioselectivity can be improved by the use of some mono or dialkylboranes instead borane itself (disiamylborane [DAB, 15,16], thexylborane [TB, 15,17], 9-BBN [15,18]). While diborane shows a 94% of regioselectivity in hydroboration of 1-hexene, this increases to 99% when using chloroborane-dimethyl suffide [15,19,20], or DAB [15,16] or thexylchloroborane-dimethyl suffide [15,21], and 99.9% if 9-BBN is used [15,18], but it's equal (94%) if TB is used [15,17]. When the substrate of the hydroboration is 2-methyl-l-butene diborane gives 99%, chloroborane-dimethyl suffide 99.5%, thexylchloroborane-dimethyl suffide 97%, and 9-BBN 99.8%. Changing the substrate to 4-methyl-2-pentene diborane gave 57%, DAB 97%, TB 66%, thexylchloroborane-dimethyl sulfide 99%, and 9-BBN 99.8%. Finally, [15] styrene as substrate gave a regioselectivity of 80% (diborane), 98% (chloroborane-dimethyl sulfide, and DAB), 95% (TB), 99% (thexylchloroborane-dimethyl sulfide), and 98.5% (9-BBN). These alkylboranes can be prepared by hydroboration of the corresponding alkene, the degree of alkylation of borane comes out from stoichiometric control [15]. See Figure 5.
Comments
The fact of adding alkyl substituents on the boron atom implies obviously an increase of the steric hindrance at the moment of interaction between the reagent and the substrate olefin. This tool makes more difficult the interaction with the most hindered carbón and accelerates the rate of attacking the less hindered sterically carbón.
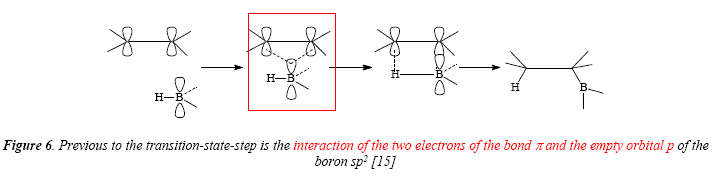
As shown above in Figures 3 and 4, the borane or its derivative in hydroboration follows a syn (cis) stereospecific addition on the alkene [15]. The transition state (T.S.) involves a four center adduct with simultaneous bonding to B and H [15]. This atoms' spatial position makes possible the formation of the new bonds B-C and H-C, as being formed in the same side of the double bond [15]. A previous to the transition-state-step is the interaction of the rwo electrons of the bond Trand the empty orbital ρ of the boron sp2, see Figure 6 [15,17].
Diborane is gaseous, but soluble in ether after dissociating into monomeric borane as etherate in varied ethers. Ether acts as catalyst in the addition reaction because of its ability of dissociating diborane [17]. The ethereal nature as solvent is not determining in the direction of addition in the hydroboration of styrene [17]. This means that the solvent does not participate actively in the transition state. On the other hand, the hydroborating agent (LiBH4 or Na BH4 or B2H6) does not make much difference in the direction of the addition either. Temperature variations didn't influence the addition direction [17]. As a general result in the distribution of boron over the double bond is from 94 to 98% in the same direction, or the less hindered carbón, in most of the cases. Indicated yields were of 90% or higher. The majority of olefins proceed in the reaction until the trialkyl borane, however, there are exceptions with the more hindered olefins that can reach the monoalkyl or dialkylborane stage [17]. The addition to styrene (20% α) or ρ-chlorosytrene (35% α) is different [17]. Styrene was chosen for studying the influence of temperature, solvent and hydroboring agent with regard to the direction of addition. A small increase in temperature produced an increase in the percentage of the α-derivative [17]. Hydroboration was also surveyed under the scope of the effect of the structure of the olefin involved in five groups, alkyl monosubstituted terminal olefins (I), alkyl disubstituted terminal olefins (II), alkyl disubstituted internal olefins (III), alkyl trisubstituted internal olefins (IV), ρ-substituted styrenes (V) [17]. The results pointed out the preferred anti-Markownikoff type of addition. Group I with alkyl substitutents, linear or branched gave 93-94% of addition of the boron to the terminal carbón. If the substituent includes somehow phenyl, the boron addition to the terminal carbón is between 80 to 90%. Group II presents a 99% of the primary alkylborane due to two substituents that exert steric compression on the more unsaturated carbón. Group III like 2- pentene and 2-hexene undergo addition of the boron in almost equal proportions to both sides of the double bond. Slight variation is seen from cis to trans isomers. Group IV shows addition results with boron added to the secondary carbón in about 98% and only 2% for the tertiary compound. Group V included results derived from the relative strong influence of the phenyl group over addition on the double bond on styrene.
Hydroboration of alkyl-substituted olefins
The directive effects of the similar structural features of different alkyl-substituted olefins during addition of hydrogen and boron to the double bond were studied [17].
Alkyl-monosubstituted terminal olefins RCH=CH2
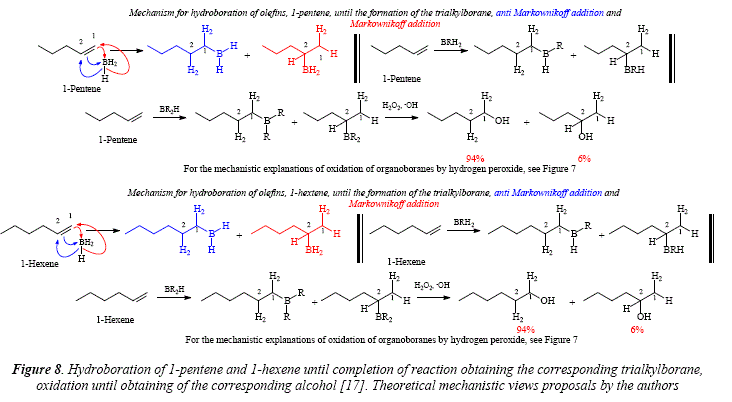
Straight-chain terminal olefins like 1-butene (Figure 7), 1-pentene and 1-hexene (Figure 8) give preferably the addition of boron to the terminal carbón in a 93-94% (6 to 7% for the secondary alkyl boron derivative) [17].

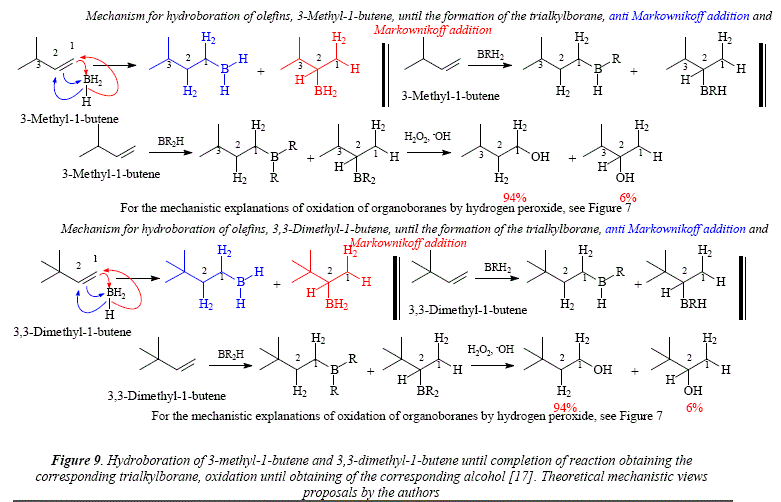
The alkyl branching makes not much difference in the boron distribution as shown with the use of 3-methyl-l-butene, 3,3-dimethyl-l-butene (Figure 9) and 4,4-dimethyl-l-pentene [17] (Figure 10).
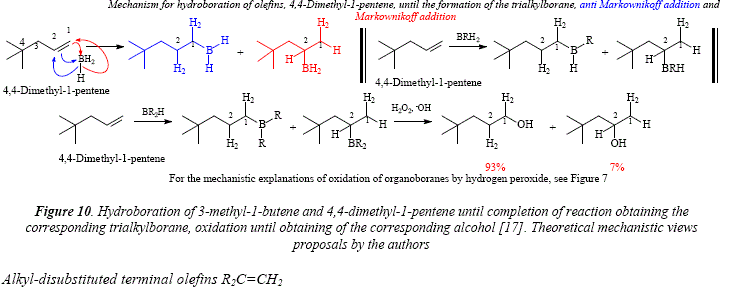
Here we are faced to two geminal alkyl groups, very hindering, by the way. This provides a terminal methylene of alkenic carbón easily approachable by voluminous groups like borane and its derivatives. The directive effect of the geminal alkyl groups (identical or not), is overpowering giving rise to boron-addition results almost exclusively on the terminal carbón. As a sample let us mention that 2-methyl-l-butene and borane give 99% of the primary alkylborane and 1% of the tertiary alkylborane, and 2,4,4-trimethyl-l-pentene [17] (Figure 11).

The immediate examples for the approach to alkyl-disubstituted internal olefins are 2-pentene and 2-hexene, which experiment hydroboration with boron addition fairly equally distributed on both extremes of the double C=C bond (Figures 12). The small difference in this distribution is observed in the geometrical isomers cis and trans.
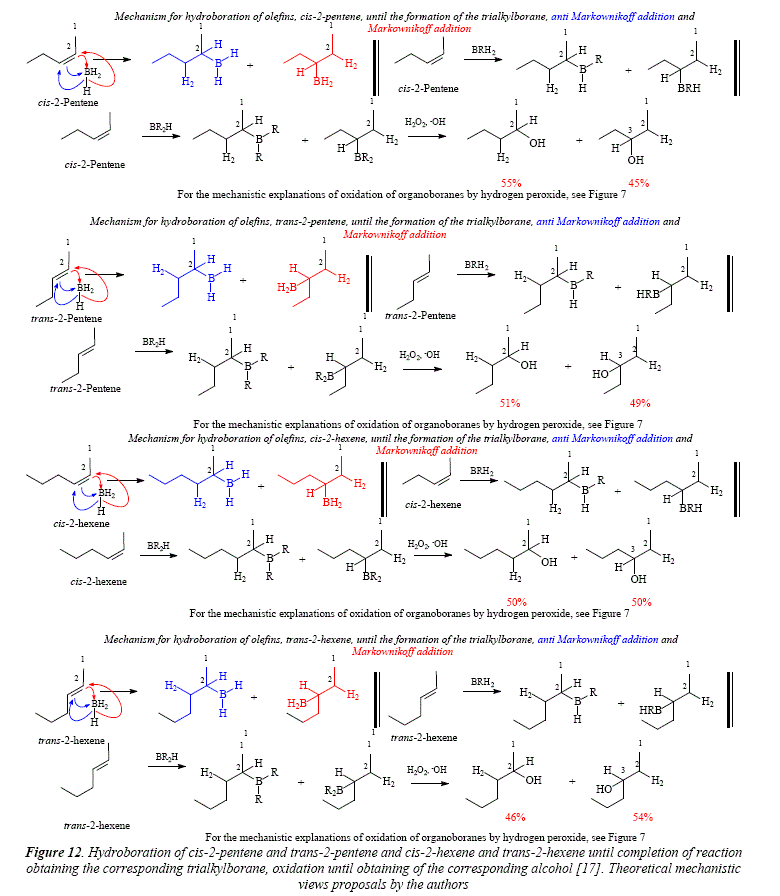
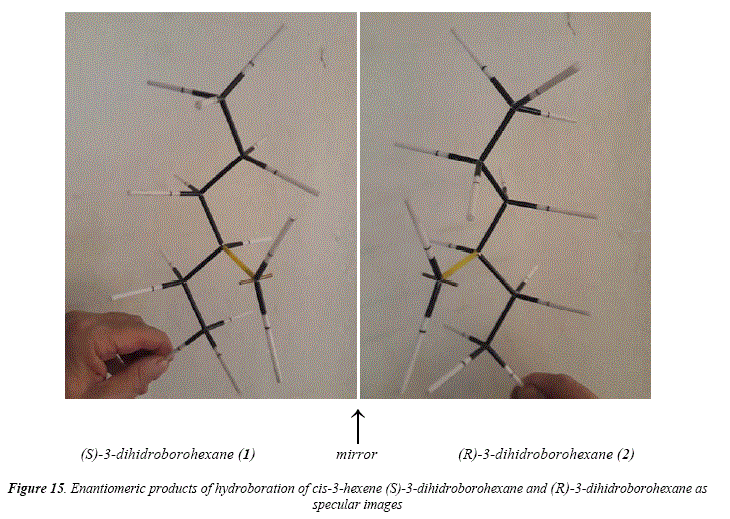
A special case is the hydroboration-oxidation of c/s-3-hexene [17], that gives a couple of enantiomeric alcohols after hydroboration and subsequent oxidation [17]. The distribution of boron on the double central C=C bond is egalitarian (50:50). The result of hydroboration and hydrogen peroxide oxidation is the enantiomer couple: (R)-3-hexanol and (5)-3-hexanol, as a racemic mixture (50:50). The mechanism for the hydroboration-oxidation of cis-3-hexene is proposed in Figure 13. In this figure it is clearly established that, being c/s-3-hexene a symmetric molecule, the alkyl side chains at each side of the double bond are identical. This means that there is no preference of the electrophile for either of the unsaturated carbons, giving a 1:1 distribution of borane. The article under current scrutiny and review [17] shows in Table IV the product of the hydroboration-oxidation of c/s-3-hexene as an only product (100%) in the column of 3-ol result. This report is not specific, meaning by this that the only product is the 3-hexanol. However, Figure 13 clearly shows the formation of a racemic mixture. Figure 14 shows through molecular models the substrate structure, c/s-3-hexene, and, the monoalkylboranes, product of the electrophilic addition, (5)-3-dihidroborohexane (1) and (R)-3-dihidroborohexane (2). Figure 15 shows the two enantiomers as specular images, demonstrating that the hydroboration gave two producís instead of only one, as Table IV [17] seems to suggest.
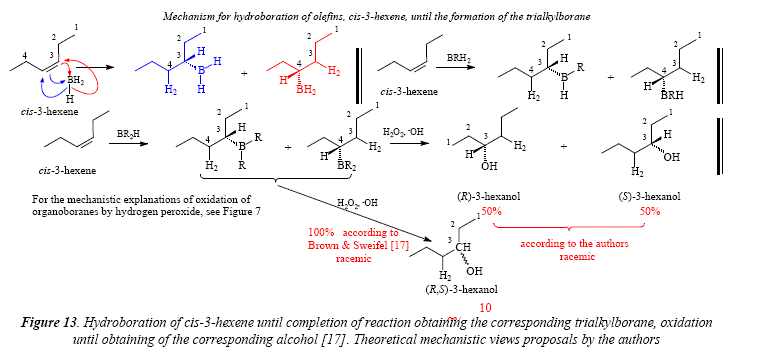
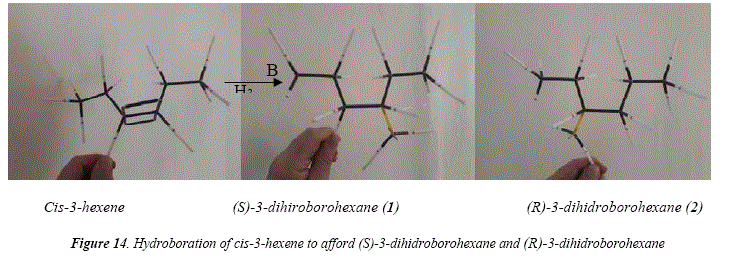
This analysis of the hydroboration of c/s-3-hexene to afford a couple of enantiomers (S)-3-dihiroborohexane (1) (R)-3-dihidroborohexane (2) as a racemic mixture as shown in Figures 14 and 15 demonstrates existence of the transition state that directs a syw (c/s) simultaneous addition of the H-B bond to the double bond. If the addition were anti (trans), it means in two instead of an only step, then the two addition-products or the 3-dihidroboranes, would be diastereomers, and not enantiomers as it's the case.
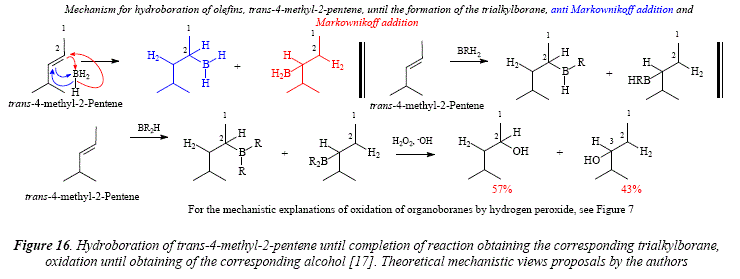
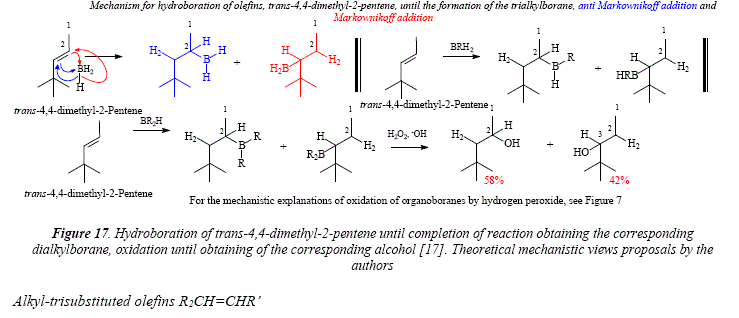
If the structural complexity at one of the side chains of the double bond is augmented, a small preference of the boron atom for the carbón atom with the less hindering side chain is manifested as shown for trans-4-methyl-2-pentene (Figure 16) and trans-4,4-dimethyl-2-pentene (Figure 17). The reaction proceeds until the dialkylborane in the case of trans-4,4-dimethyl-2-pentene [17].
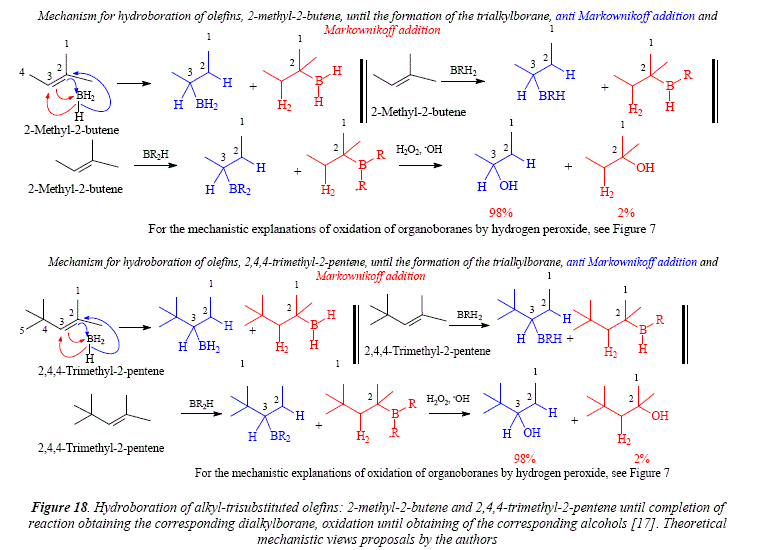
Two producís were assayed as examples of hydroboration of alkyl-trisubstituted olefins, R2CH=CHR', 2-methyl-2-butene with a 98% of accommodation of borane on the secondary unsaturated carbón and 2% on the tertiary carbón, and 2,4,4-trimethyl-2-pentene, which afforded the same distribution on the secondary and tertiary carbón as the 2-methyl-2-butene. Both compounds under mild conditions reached until the second alkylation of borane only [17].
Hydroboration of aryl-substituted olefins
Influence ofthe hydroboration (reducing) agent employed and temper'ature in the hydroboration ofstyrene
The use of one or another hydroboration agent makes no remarkable difference in the addition distribution of the couple H-B on the double bond. Thence there was no significant change being noticed with the use of LiBH4 (Et2Ü THF or diglyme), NaBH4 (diglyme) or B2H6 (diglyme) [17]. A small increase in the proportion of addition on the α-carbon of styrene was produced with increasing temperature [17].
Styrene and its derivatives
Styrene gives much different addition results of the hydroboration agent than those of alkyl-substituted olefins with proportions about 20% and 80% on the double bond [17]. See Figure 19 for a comparison of producís in different solvents and by using different hydroboration agents and at different temperatures [17].
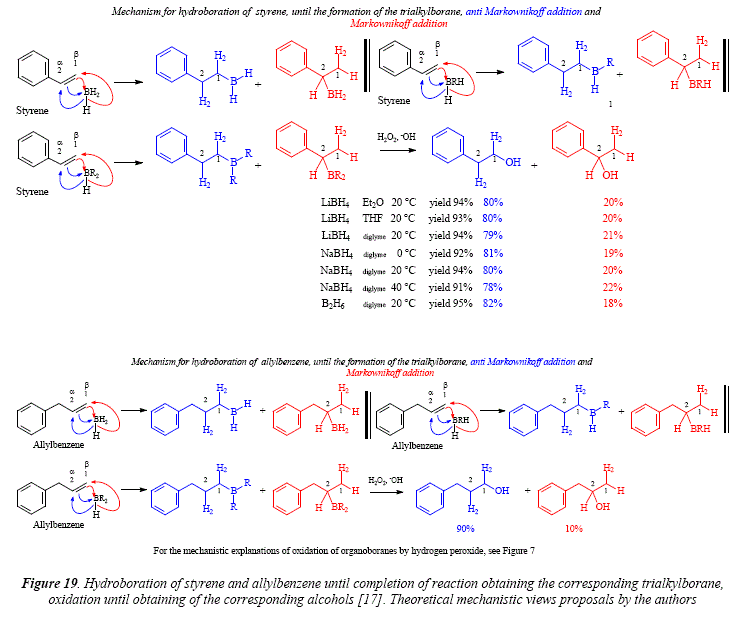
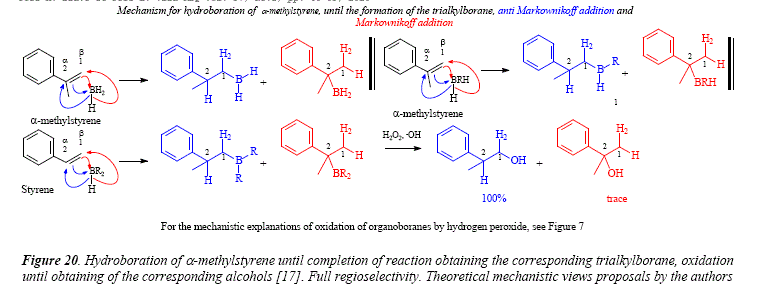
As mentioned above, the distribution is 80% at C-1 and 20% at C-2 in the hydroboration of styrene. The effect of the phenyl group is minorbut still present in allylbenzene with a 90c-1:10c-2 distribution [17], see Figure 19. The case of α-methylstyrene shows the drastic directive effect giving a full regioselectivity with al00% addition in position 1 [17]. See Figure 20.
A comparison of the directive effect of alkyl-substituted (1-pentene) vs. aryl-substituted (styrene) olefins puts in evidence that the phenyl group of styrene is less effective than alkyl groups in directing the boron atom to the terminal carbón atom. This is seen in the hydroboration of trans-1-phenylpropene, with the competition between the methyl and the phenyl group [17], Figure 21.
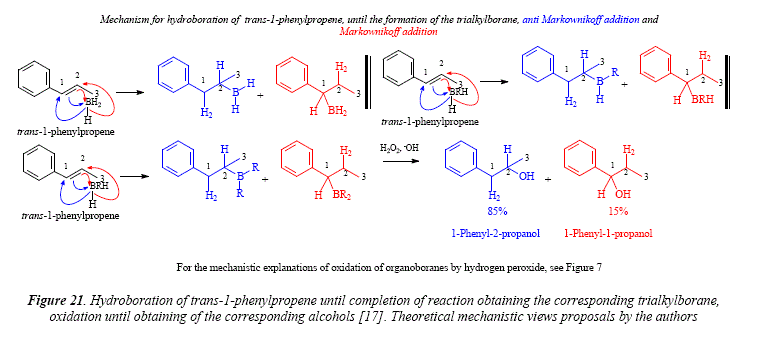
Comments
The article under current scrutiny [17], mentions a distribution of 15% for l-phenyl-2-propanol and 85% of 1-phenyl-1 -propanol, however, Figure 21 demonstrates that the distribution is exactly the opposite, namely 15% for 1-phenyl-1 -propanol and 85% of l-phenyl-2-propanol. This is an error of Brown and Zweifel [17].
p-Substituted styrenes
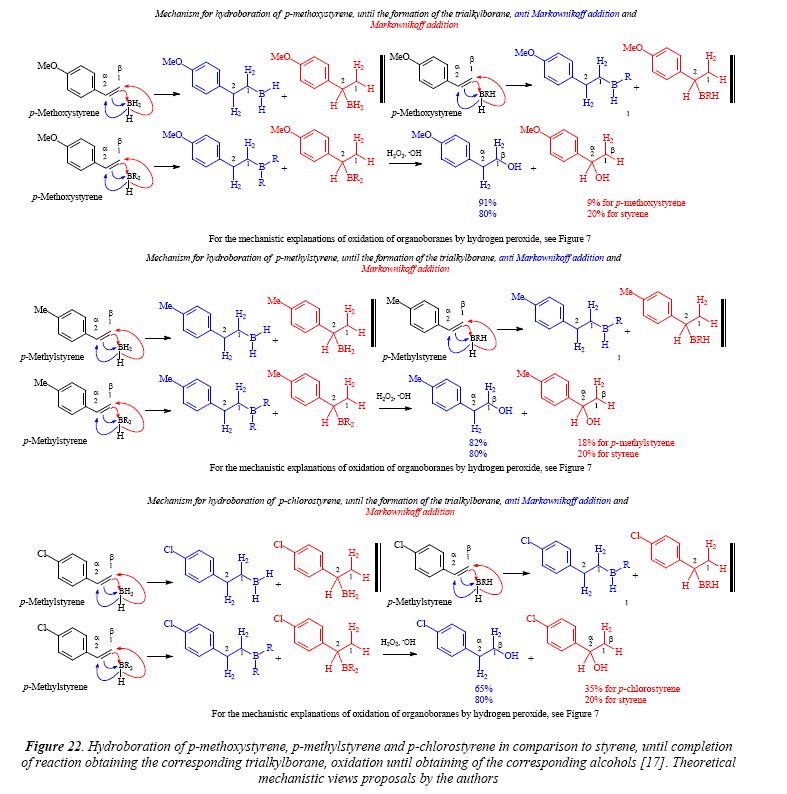
Due to the remarkable influence of the phenyl group in styrene on the tendency for the diborane when choosing a carbón atom over the other in the double bond, incited to the investigation of styrenes substituted in para [17]. This study was hoped to give enlightenment about the nature of the forces controlling the preference in the addition reaction.
The methyl substituent in styrene, provoked a minor effect, with a diminution from 20% in styrene in the a carbón to 18% in the para derivative. The methoxy substituent exerted a higher effect diminishing form 20% in the a position in styrene to 9% in the para derivative. Finally, the p-chloro substituent augments the distribution in the a position of the double bond until 35% from 20% in styrene [17], Figure 22.
CONCLUSIÓN
The direction of addition of borane on olefins exposed in the examples above has been examined through the corresponding alcohols after oxidation [171. The hydroboration repeats three times, each time with a new molecule of substrate until reaching the trialkylated boron stage [17]. There is no evidence that the final distribution signaled by the alcohols is the same at each stage in the borane's alkylation pathway [17]. But, authors consider the distributions of the alcohols derived, as the same preferred by borane in the each of the preceding steps in the addition to olefins. Most of olefins proceeds to the trialkylborane R3B [17]. Other olefins, in function of the alkyl increasing structural complexity arrive to the dialkylborane stage or the monoalkylborane stage [17]. All that have been exposed until here, regards with the directive effects derived from steric effects on the direction of addition of borane on olefins [17]. However, authors signaled that steric directive effects are not alone in exerting influence in the addition of borane on olefins [17]. Some of them being much different in branching, but with similar effect in distribution in results of the electrophilic addition, as seen in the foliowing table [ 17].
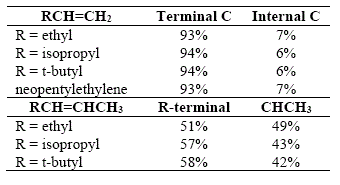
All data collected and its previous analyses require an explanation other than the steric effects, and it concerns the electronic influences clearly playing an important role in controlling the direction of addition of the B-H bond to thedoublebond[17].
As shown above (figures 3 and 4), the addition of diborane to olefins happens clearly in a cis manner (see also figures 13-15), involving a four-center transition state [17]. The B-H bond is polarized with the hydrogen possessing a hydride character. Henee, by analogy, the electronic shifts are like in the addition of H-Cl to double bonds. See Figure 23, where borane adds to propylene, that kind of electronic shift is the explanation for the addition of boron to the terminal carbón. The same explanation is applicable to 2,2-dialkylethylenes (99% C-l) and for the secondary position in trisubstituted olefins (98%).
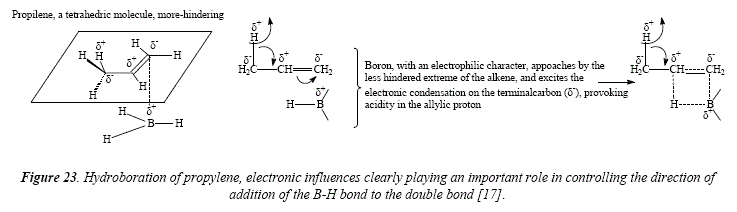
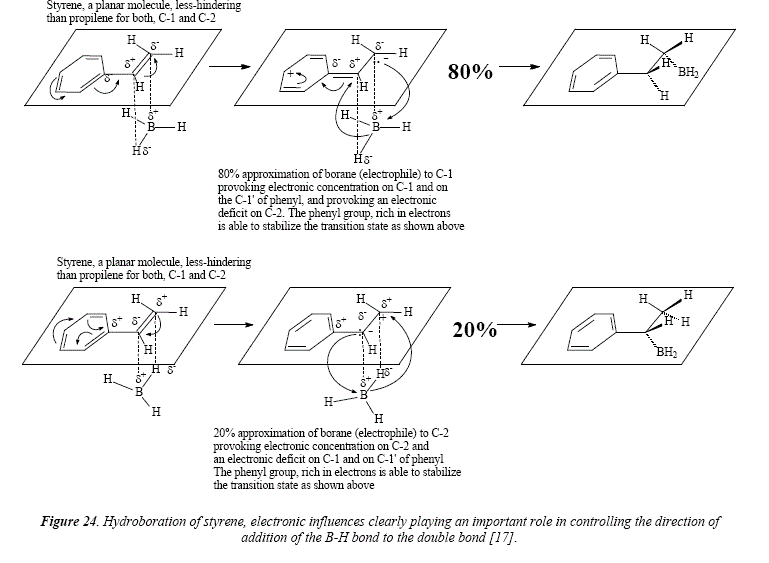
A similar approach can be employed for styrene to explain the preference of the boron atom for the ending position in the side chain. This approach also let explain the higher preference of C-2 with respect of the similar carbón in alkyl-substituted olefins: "In order to account for the enhanced substitution which occurs in the α-position, we must recognize that the phenyl group can also stabilize a negative charge in the α-position as in the benzyl anión. This transition state would be stabilized by an electron-withdrawing substituent, such as ρ-chloro-, and rendered less stable by an electrón supplying substituent, such as ρ-methoxy" [17]. See Figure 24. The transition state at 80% possesses three resonant structures (two of them corresponding to benzene), in contrast to transition state at 20% that dispose of only 2 resonant structures (corresponding to benzene), being thus less stable.
REFERENCES
1. Bravo, J. 2005, The organic chemistry notebook series, a didactical approach. Theoretical mechanistic approach to diasteroselective synthesis of cis-l,2-dialkenylcyclopropanols and subsequent oxy-Cope rearrangement by Jin Kun Cha et al, Rev. Bol. Quim., 23 (1), 1-10. [ Links ]
2. Bravo, J.A., Mollinedo, P., Peñarrieta, J.M., Vila, J.L. 2013, Mechanistic views of intramolecular hydroxycyclopropanation of ovinyl carboxylic esters, Rev. Bol. Quim., 30 (1), 24-41. [ Links ]
3. Bravo, J.A., Vila, J.L. 2014, Mechanistic views of stereoselective synthesis of tri and tetra-substituted alkenes, part I; the organic chemistry notebook series, a didactical approach, n° 3. Rev. Bol. Quim., 31 (1), 61-67. [ Links ]
4. Bravo, J.A., Vila, J.L. 2015, Mechanistic views of stereoselective synthesis of tri and tetra-substituted alkenes, part II; the organic chemistry notebook series, a didactical approach, n° 4, Rev. Bol. Quim., 32 (1), 15-23. [ Links ]
5. Vila, J.L., Bravo, J.A. 2015, Synthesis of alkenes by fragmentation reactions; Mechanistic views; the organic chemistry notebook series, a didactical approach, n° 5, Rev. Bol. Quim., 32 (2), 37-44. [ Links ]
6. Bravo, J.A., Vila, J.L. 2015, Synthesis of alkenes by oxidative decarboxylation of carboxylic acids; Mechanistic views; the organic chemistry notebook series, a didactical approach, n° 6, Rev. Bol. Quim., 32 (3), 45-52. [ Links ]
7. Bravo, J.A., Vila, J.L. 2015, Synthesis of alkenes from ketones via arylsulphonyl-hydrazones; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 7, Rev. Bol. Quim., 32 (4), 82-89. [ Links ]
8. Bravo, J.A., Vila, J.L. 2015, Stereospecific synthesis of alkenes from 1,2-diols; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 8, Rev. Bol. Quim., 32 (5), 121-125. [ Links ]
9. Bravo, J.A., Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part I; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 9, Rev. Bol. Quim., 33 (1), 27-33. [ Links ]
10. Bravo, J.A., Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part II; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 10, Rev. Bol. Quim., 33 (2), 95-103. [ Links ]
11. Bravo, J.A., Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part III; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 11, Rev. Bol. Quim., 33 (3), 127-133. [ Links ]
12. Bravo, J.A., Vila, J.L. 2017, Claisen rearrangement of allyl vinyl ethers to afford alkenes, part IV; mechanistic theoretical proposals; the organic chemistry notebook series, a didactical approach, n° 12, Rev. Bol. Quiñi., 34 (2), 40-49. [ Links ]
13. Bravo, J.A., Vila, J.L. 2017, Mechanistic theoretical proposals for: alkenes by claisen rearrangement of alfa-allylthio carbenes; aza-cope rearrangement of 4-butenyliminium ions; 2-substituted pyrrolidine derivatives; synthesis of perhydrogephyrotoxin, key step; part v; the organic chemistry notebook, n° 13, Rev. Bol. Quiñi., 34 (5), 142-149. [ Links ]
14. Bravo, J.A., Vila, J.L. 2017, Obtaining of alkenes by reductive coupling of carbonylic compounds; synthesis of z,e-6-dodecene, syntheses of flexibilene and isocaryophyllene, mechanistic views; the organic chemistry notebook, n° 14, Rev. Bol. Quiñi., 35 (3), 73-84. [ Links ]
15. Carey, F.A., Sundberg, R.J. Advanced Organic Chemistry, Part B: Reaction and Synthesis, Plenum Press, 3ri ed., 1991, New York, U.S., pp. 200-205. [ Links ]
16. Zweifel, G., Brown, H.C. 1963, Hydration of olefins, dienes, and acetylenes via hydroboration, Org. React. of Chemistry, 13, 1-53. [ Links ]
17. Brown, H.C, Zweifel, G. 1960, Hydroboration. VIL Directive effects inthe hydroboration of olefins, J. Am. Chem. Soc, 82(17), 4708-4712. [ Links ]
18. Brown, H.C, Knights, E.F., Scouten, C.G. 1974, Hydroboration. XXXVI. Direct route to 9-borabicyclo[3.3.1]nonane via the cyclic hydroboration of 1,5-cyclooctadiene. 9-Borabicyclo[3.3.1]nonane as a uniquely selective reagent for the hydroboration of olefins, J. Am. Chem. Soc. 1974 96(25), 7765-7770. [ Links ]
19. Brown, H.C, Ravindran, N., Kulkarni, S.U. 1979, Hvdroboration. 52. Monohaloborane-methvl sulfide adducts as new reagents for the hvdroboration of alkenes. A convenient svnthesis of dialkvlhaloboranes and their derivatives for organic svnthesis. J. Org. Chem., 44(14), 2417-2422. [ Links ]
20. Brown, H.C, Racherla, U.S. 1986, Hydroboration. 76. Revisión of the regioselectivity of the hydroboration of alkenes with dihaloborane-dimethyl sulfide complexes, J. Org. Chem., 51(6), 895-897. [ Links ]
21. Brown, H.C, Sikorski, J.A., N., Kulkarni, S.U., Lee, H.D. 1980, Thexylchloroborane-methyl sulfide. A selective monohydroborating agent with exceptional regioselectivity, J. Org. Chem., 45(22), 4540-4542. [ Links ]

