











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkRevista Boliviana de Química
versión On-line ISSN 0250-5460
Rev. Bol. Quim vol.36 no.3 La Paz ago. 2019
ARTÍCULOS ORIGINALES COMPLETOS
Reacciones primarias de abstracción de hidrógeno del trans-1,3-butadieno mediante radical hidroxilo
Primary reactions of hydrogen abstraction from trans-1,3-butadiene by using radical hydroxyl
Rolvideer J. González Herrera1, *, Karen R. Martínez-Ramírez1, Marcos A. Franco Frings1
1 Departamento de Física y Química. Facultad de Ciencias Exactas y Naturales FACEN, Universidad Nacional de Asunción UNA, P.O. Box 1039-1804, Ruta Mcal. Estigarribia, Km 11, Tel. +59521-585602, Campus Universitario UNA-San Lorenzo, Paraguay., www.facen.una.py
*Corresponding author: rolvideeriavier@gmail.com
Received: 06/02/2019 Accepted: 08/16/2019 Published: 08/30/2019
Resumen
Se han investigado en forma teórica los canales de abstracción de los hidrógenos del 1,3-butadieno (C4H6) por hidroxilo radical, en su estado electrónico fundamental, en fase gaseosa y en condiciones normales de presión y temperatura.
Se realizó un estudio de los rotámeros del C4H6, habiéndose encontrado que el confórmero trans es 12,3 kJ/mol más estable que la forma cis. Se determinaron tres canales distintos de abstracción de hidrógenos del C4H6, debido a la presencia de sus tres protones químicamente no equivalentes. Se caracterizaron todas las estructuras moleculares involucradas en cada canal reactivo, confirmados por sus frecuencias vibracionales, coordenada intrínseca de reacción (IRC) y vectores de transición.
Se utilizó el método del funcional de la densidad (DFT), con el funcional híbrido B3LYP y base de Dunning aug-cc-pVTZ, incorporados en el paquete Gaussian 03. En todos los casos se procedió a extrapolar la energía al límite de base completa (CBS) utilizando un método exponencial y aplicando correcciones de la energía del punto cero (ZPE).
Se reportan energías de activación negativas para los tres canales de reacción y la formación de complejos posreactivos.
Palabras clave: Butadieno, extracción de hidrógeno, DFT, estudio teórico.
Abstract
The abstraction channels of 1,3-butadiene (C4H6) hydrogens by radical hydroxyl have been theoretically investigated, in their fundamental electronic state, in the gas phase and under normal conditions of pressure and temperature.
A study of the C4H6 rotamers was performed, having found that the trans conformer is 12,3 kJ / mol more stable than the cis forra Three different C4H6 hydrogen abstraction channels were determined, due to the presence of its three chemically non-equivalent protons. All molecular structures involved in each reactive channel were characterized, confirmed by their vibrational frequencies, intrinsic reaction coordinate (IRC) and transition vectors.
The density functional method (DFT) was used, with the hybrid functional B3LYP and Dunning base aug-cc-pVTZ, incorporated in the Gaussian 03 package. In all cases the energy was extrapolated to the base limit Complete (CBS) using a exponential method and applying zero point energy corrections (ZPE).
Negative activation energies are reported for the three reaction channels and the formation of post-reactive complexes.
Keywords: Butadiene, hydrogen abstraction, conformers, DFT, theoretical study.
INTRODUCCIÓN
Los compuestos orgánicos volátiles (COVs) se emiten a la atmósfera a partir de fuentes antropogénicas y biogénicas, como también pueden formarse in situ como producto de las transformaciones atmosféricas de otros COVs [1].
Uno de estos compuestos es el butadieno, sustancia gaseosa ampliamente utilizada por las industrias en la fabricación de polímeros y otros subproductos derivados del petróleo [2], emitido al ambiente por escapes de automóviles [3] y quema de neumáticos en desuso[4,5].
Además, este COV está catalogado como sustancia altamente cancerígena [6-9], capaz de interaccionar químicamente con oxidantes atmosféricos [10,11] y producir sustancias que contribuyen al smog fotoquímico [12-14].
Durante mucho tiempo, la caracterización de la estructura conformacional (posibles rotámeros) del butadieno, representó un tema de gran interés para la comunidad científica. Esto, debido a que representa al sistema más simple que posee una conjugación de enlaces dobles y por ser una molécula de interés medioambiental [1,2].
El primer trabajo experimental que reporta la estructura conformacional del 1,3-Butadieno data de 1939, mediante técnicas de difracción de electrones, innovador para la época [15]. Las fotografías obtenidas fueron difusas, por lo que la determinación de la conformación de la molécula se realizó con dificultad y según los propios autores, sin la precisión deseada.
Más tarde, otros investigadores abordaron nuevamente la caracterización estructural del 1,3-butadieno aprovechando las mejoras en las técnicas de difracción de electrones, obteniendo resultados más fiables. Reportaron la caracterización de la forma trans de la molécula, que adopta una configuración plana, y no detectaron la presencia de otros confórmeros [16].
Uno de los primeros trabajos teóricos sobre la conformación estructural de moléculas data de 1970, aplicando cálculos de Hartree-Fock con orbitales tipo Slater (STO) para investigar las barreras torsionales -C-C- de algunos cationes alquílicos [17]. El mismo determinó la presencia de otro confórmero estable, que, según los resultados, adoptaba una conformación cis planar y mostró la posibilidad de que las rotaciones de los grupos moleculares alrededor de enlaces simples carbono-carbono generen otros confórmeros estables adicionales para cada molécula.
Otro trabajo posterior, sobre las posibles estructuras de equilibrio adoptadas por el butadieno data del año 1976, en el cual se aplica el método de fuerza de Pulay (ab initio) con un conjunto de bases A(7,3) y se lograron caracterizar cuatro confórmeros rotacionales. Entre estos confórmeros la configuración trans resultó ser la de menor energía, sin embargo, según los propios autores, la cuestión de si el segundo confórmero estable es cis o gauche, aún no quedaba confirmada [18].
Luego de estas investigaciones, han aparecido numerosos estudios teóricos sobre barreras torsionales, frecuencias vibratorias y conformaciones estructurales del 1,3-butadieno [19-22]. En todos ellos, los autores coinciden en que la estructura estable de mínima energía, es una molécula plana de conformación trans y que el segundo confórmero estable adopta la conformación gauche no planar. Además, se pone de manifiesto la necesidad de utilizar geometrías optimizadas en cada nivel de cálculo para inferir resultados fiables y no utilizar parámetros estructurales experimentales, ya que pequeñas variaciones en la geometría estructural de la molécula, puede derivar en grandes diferencias energéticas.
Los estudios dedicados a las reacciones de los COVs con los distintos oxidantes atmosféricos, indican que la principal vía de degradación es la reacción con radical hidroxilo (OH•), siendo este radical un importante intermediario reactivo en la química atmosférica [1,23]. Si la concentración de estos radicales disminuyera debido al incremento de relativa oscuridad (invierno, horas de la tarde) y la concentración de ozono aumentase por algún episodio de contaminación, la reacción con ozono puede ser una importante vía de degradación. Además, en las zonas donde las concentraciones de átomos de C1- son significativamente mayores que la concentración promedio global, tal como podría ocurrir en el aire costero urbano y algunas regiones industriales, especialmente por las mañanas, sus efectos oxidativos son considerables [24].
Las reacciones de adición del OH• a los enlaces dobles del 1,3-butadieno ya fueron ampliamente abordadas por la comunidad científica [25-27], no así las reacciones de abstracción de hidrógeno. Los procesos de adición prevalecen sobre las reacciones de abstracción, debido a la alta densidad electrónica alrededor de los enlaces dobles >C=C< [1]. No obstante, también se reportan investigaciones donde se afirma que las reacciones de abstracción de hidrógenos pueden ser vías muy importantes en la degradación de los COVs en la atmósfera [28,29].
Las reacciones de los radicales OH• con las olefinas, incluido el butadieno, pueden presentar energías de activación negativas [28]. Estas energías negativas sugieren la formación de complejos de Van der Waals, difíciles de caracterizar experimentalmente, no así computacionalmente.
Li et al. [26], realizaron un estudio experimental y teórico de los canales de reacción del OH• con 1,3-butadieno. Aplicaron técnicas de velocidad relativas para determinar las constantes de velocidad de la reacción y lo contrastaron aplicando métodos teóricos. Afirman que la mejor estimación de la energía relativa se basa en un cálculo de un solo punto en el nivel de teoría MP4/6-311++G(dp,d), más correcciones de ZPE utilizando la geometría optimizada en el nivel de teoría MP2/ 6-311++G(d,p) [30]. Lograron caracterizar mínimos y estados de transición de los procesos de adición y abstracción, pero no se reporta formación de complejos de Van der Waals.
Según nuestra revisión bibliográfica y hasta la fecha, no se ha reportado previamente la formación de complejos en las reacciones de abstracción de hidrógenos del 1,3-butadieno por radicales OH•.
RESULTADOS Y DISCUSIÓN
Se realizó una revisión de los rotámeros del 1,3-butadieno aplicando la rutina SCAN implementada en el Gaussian 03. Para esto se utilizaron 72 rotaciones de 5° de los grupos moleculares que conforman la molécula, unidos por el enlace simple =CC=.
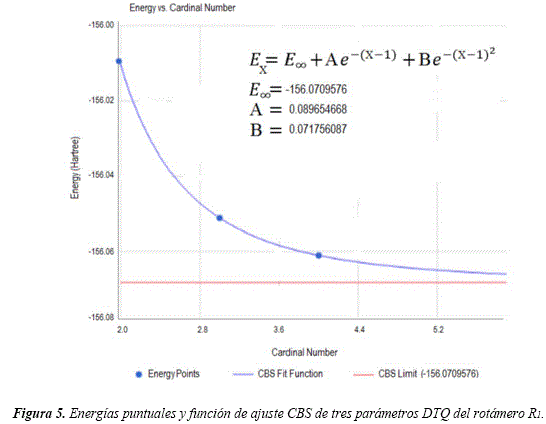
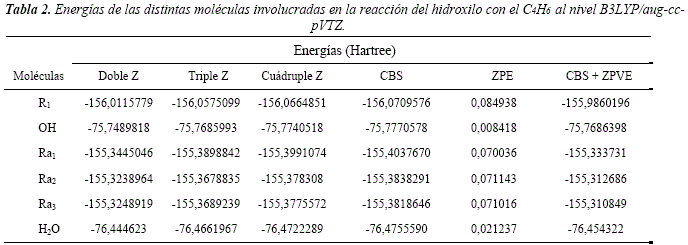
Para la optimización completa de las geometrías y cálculos de energía, se utilizó el método DFT con el funcional híbrido B3LYP [31,32] y base de Dunning aug-cc-pVTZ (B3LYP/aug-cc-pVTZ// B3LYP/aug-cc-pVTZ) [33]. Todos los cálculos se llevaron a cabo utilizando el paquete de programas Gaussian 03 [34], con el auxilio del visualizador gráfico Gauss View 4.1. En todos los casos se realizaron los cálculos de las frecuencias armónicas vibracionales, utilizando la rutina frequency. Además, se caracterizaron las energías del punto cero (ZPE) y se verificó que las frecuencias sean todas reales para los mínimos y una sola imaginaria para los estados de transición. Se aplicaron correcciones adicionales a la energía, realizando extrapolación a base infinita (CBS, siglas en inglés). Se optó por utilizar un método exponencial mixto de tres parámetros para la convergencia de la energía de correlación, por proporcionar buenos resultados según se reporta en la literatura [21-35]. Este método, también denominado de tres parámetros, desarrollado por Peterson et al. [36], utiliza la relación:
![]()
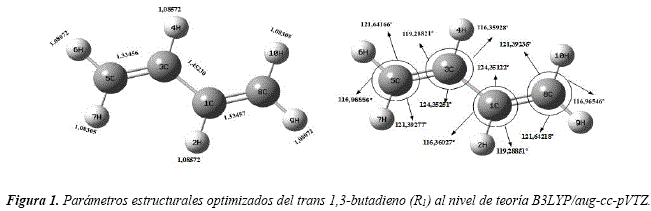
Esta ecuación ya está incorporada en la calculadora online CBS [37], con la cual fueron determinadas las energías extrapoladas al límite de base completa. La conformación espacial adoptada por el confórmero inicial R1 del 1,3-butadieno, caracterizando sus distancias interátomicas (en angstróm) y ángulos de enlace (en grados), se puede observar en la figura 1.
Los carbonos e hidrógenos que conforman la molécula subyacen todos en un solo plano, según se puede observar en la Figura 2, y verificando sus ángulos diedros.
Los parámetros estructurales obtenidos para la molécula inicial R1, presentan buena concordancia con los datos experimentales, obtenidos mediante técnicas de difracción de electrones, reportados por Schomaker et al. [15] y Almennmgen, et al. [16], que se pueden observar en la Tabla 1.
Se identificaron dos conformaciones estables adicionales a partir de la molécula inicial R1. La rotación del ángulo diedro D(C5 = C3C1= C8), tomando como eje de rotación al enlace simple C3C1, conduce a la formación de los rotámeros R2 y R3, a los 150° y 210° de rotación, respectivamente. La Figura 3 presenta la conformación espacial adoptada por ambos rotámeros. Se observa que estas moléculas no son planas, con ángulo diedro C=C-C=C de ~ 32° y que una es aproximadamente la imagen especular de la otra.
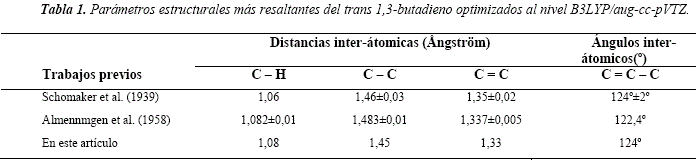
Ambos rotámeros poseen energías esencialmente iguales y son menos estables que el rotámero Ri, en unos 12,3 kJ/mol. Este resultado está en excelente acuerdo con los 12,4 kJ/mol reportado por Feller et al. [22], realizados con métodos ab initio de alto nivel, utilizando la teoría de clústers acoplados con excitaciones triples cuasiperturbativas. También está en buen acuerdo con lo reportado por Sancho-García et al. [21], donde mediante cálculos ab initio al nivel HF/cc-pVTZ, CCSD(T)/cc-pVTDZ y MP2/cc-pVTZ informan diferencia de energía de 12,76 kJ/mol para los dos primeros métodos y 12,84 kJ/mol para el último.
De igual manera, Skaarup et al. [18], reportan diferencias de energía de 11,97 kJ/mol por el método de fuerza de Pulay (ab initio) con un conjunto de bases A(7,3) y Murcko, et al. [20], reportan diferencias energéticas de 12,13 kJ/mol y 13,39 kJ/mol por el método G2 y CBS-Q, respectivamente para los rotámeros.
Al aplicar la estadística de Maxwell-Boltzmann, se obtiene que la razón entre la abundancia de la molécula Ri y los otros dos rotámeros es ~ 99:1 [38]. La interconversión entre los rotámeros R2 y R3 requiere superar una barrera energética muy baja, de aproximadamente 1,4 kJ/mol que está en buen acuerdo con los 2 kJ/mol reportado por Feller etal. [22].
Partiendo de las geometrías optimizadas al nivel de teoría B3LYP/aug-cc-pVTZ, se realizaron los cálculos puntuales de energía necesarios para la corrección a base infinita con las bases aug-cc-pVNZ (N = D y Q).
El rotámero de menor energía denominado R1, fue utilizado para realizar el estudio de las reacciones de abstracción de hidrógenos y la optimización de las estructuras de los radicales asociados. Las reacciones de abstracción de hidrógenos del 1,3-butadieno por el OH*, siguen la reacción:
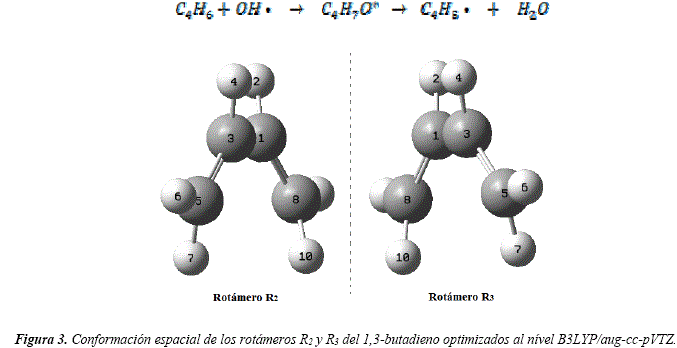
Se caracterizaron tres radicales a partir de la abstracción de sus hidrógenos. Los pares de hidrógenos: H2 y H4, H6 y H9, H7 y H10 resultaron ser equivalentes y condujeron a la formación de los radicales Ra1, Ra2 y Ra3, respectivamente. Las disposiciones espaciales y configuraciones estructurales de dichos radicales obtenidos por abstracción de los hidrógenos se observan en la Figura 4.
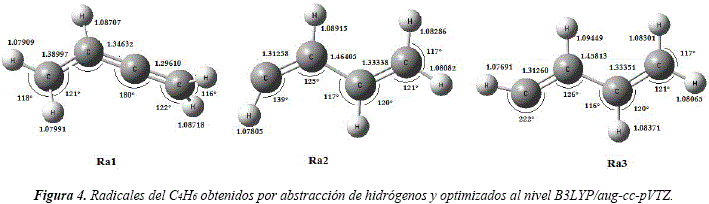
En la Tabla 2 se detalla las energías del confórmero inicial R1, del hidroxilo, de los radicales y del agua que se forma en la reacción de abstracción de hidrógenos. También se incluye las energías CBS obtenidas de las proyecciones a base infinita, siendo presentada en la Figura 5 una de estas extrapolaciones, la correspondiente al rotámero R1. Entre los radicales obtenidos por abstracción de hidrógenos, el de mayor estabilidad es el radical Ra1 con una diferencia energética de 55,25 kJ/mol y 60,08 kJ/mol, con respecto a las energías de los radicales Ra2, Ra3, respectivamente. Esto permite concluir que el canal reactivo que conduce a la formación de este radical es termodinámicamente el más favorecido.
Estos resultados se hallan en buen acuerdo con lo reportado por Parker et al. [39], donde informan que el radical más estable es el Ra1 con diferencias energéticas de 57,6 kJ/ mol y 60,4 kJ/mol con respecto a Ra2 y Ra3, respectivamente, realizando cálculos al nivel B3LYP/6-311G(d,p).
Esta relativa estabilidad del radical Ra1, con respecto a los otros radicales, se debe a la formación de enlaces dobles acumulados y una deslocalización del electrón desapareado (Ver Figura 4). Con respecto a la molécula inicial R1, este radical presenta una energía relativa de 1712,58 kJ/mol.
Cada canal de abstracción se produce previa formación de una estructura inestable denominada estructura del estado de transición (TS), cuyas disposiciones conformacionales se observan en la Figura 6. Para la determinación de dichas estructuras, se optó por utilizar el método BERNY incorporado en el paquete Gaussian 03. La validez de estas estructuras fue verificada a través de sus frecuencias vibracionales, vectores de transición y coordenadas intrínsecas de reacción (IRC).
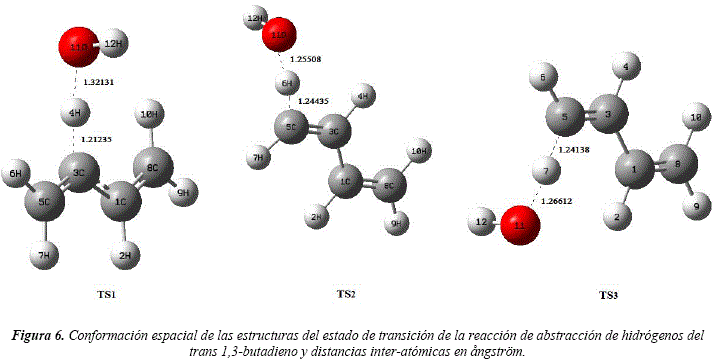
La molécula denominada TS1 se obtiene del canal de abstracción del H2 y H4. De la abstracción del H6 y H9 se obtiene la molécula TS2. La molécula TS3 se obtiene por el canal de abstracción del H7 y H10.
Estas estructuras presentan una sola frecuencia armónica vibracional negativa y los IRC confirman que estas estructuras conectan los reactivos con los productos. El IRC correspondiente al canal de reacción del TS1 se puede observar en la Figura 7.
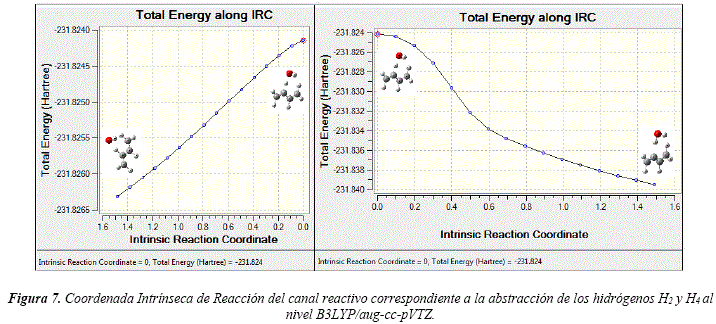
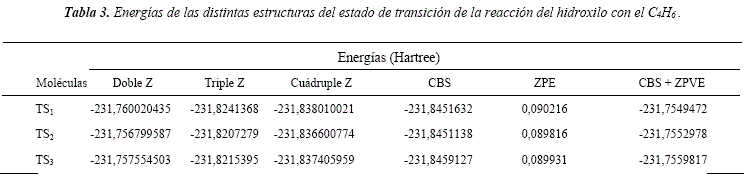
Las energías de los TS para los distintos canales de abstracción de hidrógenos se detallan en la Tabla 3. Comparando las energías de las estructuras correspondientes a los estados de transición con las energías de los reactivos, se determina que los canales de abstracción poseen energías de activación negativas. Estas energías de activación negativas sugieren la formación de complejos Van der Waals pre y pos reactivos.
Li et al. [26], reporta dos estructuras del estado de transición para el proceso de abstracción de hidrógenos, con barreras energéticas de 17,57 kJ/mol coincidente con nuestro primer canal de abstracción y 28,45 kJ/mol para el segundo y tercer canal de abstracción. En este trabajo no se reportan formación de complejos de Van der Waals.
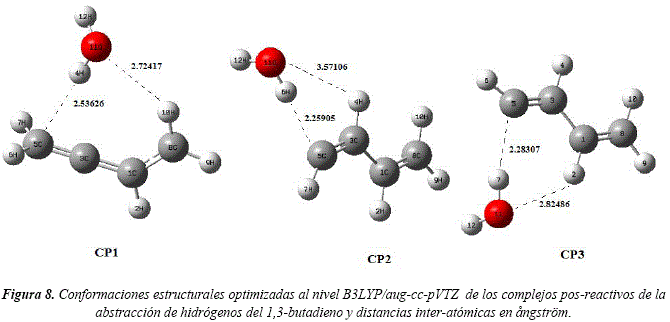
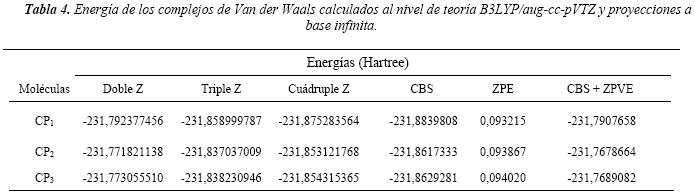
La optimización completa de las geometrías derivadas del último punto del IRC, condujeron a la identificación de tres complejos pos reactivos, denominados complejos productos CP1, CP2 y CP3, correspondiente a los tres canales de abstracción estudiados. Lastimosamente todos los intentos por obtener estructuras optimizadas correspondientes a los complejos pre-reactivos fueron infructuosos. La Figura 8 muestra las conformaciones estructurales de los complejos caracterizados y sus energías se detallan en la Tabla 4.
El complejo CP1 presenta menor energía que los demás complejos, en concordancia con la gran estabilidad del radical Ra1 procedente de dicho canal de abstracción. Todos los canales presentan reacciones sin barrera y el perfil de reacción se puede observar en la Figura 9.
En la Tabla 5 se presentan las frecuencias vibracionales de dichos complejos y de todas las estructuras implicadas en los distintos canales reactivos de abstracción de hidrógenos.
CONCLUSIONES
Se ha caracterizado tres rotámeros del 1,3-butadieno, del cual la conformación trans es la de menor energía. Este rotámero es 12,3 kJ/mol más estable que los otros rotámeros, lo cual le confiere una abundancia relativa superior al 99% a temperatura ambiente. Debido a la baja abundancia atmosférica del confórmero cis, los productos derivados de las reacciones de los oxidantes troposféricos con esta molécula serían mínimos.
Además, se han identificado tres canales de reacción de abstracción de hidrógenos que presentan energías de activación negativas. Este hecho motivó la búsqueda y determinación de complejos de Van der Waals asociados a cada reacción. No fue posible caracterizar complejos pre-reactivos, no obstante, se identificaron los complejos posreactivos de los tres canales de reacción. El complejo CPi presenta mayor estabilidad que los complejos CP2 y CP3 en unos 55,25 kJ/mol y 60,07 kJ/mol, respectivamente.
La gran estabilidad del complejo CP1 muestra que el canal de abstracción de los hidrógenos H2 y H4 se encuentra favorecido termodinámicamente, por lo que el radical Ra1 es el producto termodinámico de la reacción de abstracción.
Según nuestra revisión bibliográfica y hasta la fecha, es el primer reporte sobre la formación de complejos en las reacciones de abstracción de hidrógenos del 1,3-butadieno por radicales OH•.


REFERENCIAS
1. Atkinson, R. 2003, Kinetics of the gas-phase reactions of OH radicals with alkanes and cycloalkanes, Atmos. Chem. Phys., 3, 2233-2307. [ Links ]
2. Klaus, W., Hans-Jurgen, A. Química Orgánica Industrial. Ed Reverte, 1981, Barcelona, España, pp. 415.
3. Baker, J., Are, J. Atkinson, R. 2005, Formation and Reaction of Hydroxycarbonyls from the Reaction of OH Radicals with 1,3-Butadiene and Isoprene, Environ. Sci. Technol, 39(11), 4091-4099. [ Links ]
4. Reisman, J. 1997, Air emissions from scrap tire combustion, United States Environmental Protection Agency, Washington D.C., United States, 117. [ Links ]
5. Duarte, S., Velázquez, E., Dullak, A. 2016, Pirolisis convencional de neumáticos en desuso para la producción de combustibles líquidos, Revista Científica de la UCSA, 3(2), 25-32. [ Links ]
6. Cote, L., Bayard, S. 1990, Cancer Risk Assessment of 1,3-butadiene, Environmental Health Perspectives, 86, 149-153. [ Links ]
7. Ye, Y., Galbally, I., Weeks, I. 1997, Emission of 1,3-butadiene from petrol-driven motor vehicles, Atmospheric Environment. 31(8), 1157-1165. [ Links ]
8. Gustafson, P., Barregard, L., Strandberg, B., Sallsten, G. 2007, The impact of domestic wood burning on personal, indoor and outdoor levels of 1,3-butadiene, benzene, formaldheyde and acetaldehyde, Journal of Environmental Monitoring, 9, 23-32. [ Links ]
9. Luttrell, W. 2013, 1,3-Butadiene, Journal of Chemical Health & Safety, 54-56. [ Links ]
10. Cvetanovic, R., Doyle, L. 1960, Reaction of oxygen atoms with butadiene, Can J. Chem, 38, 2187-2195. [ Links ]
11. Haller, I., Srinivasan, R. 1964, Photochemistry of 1,3-butadiene: Details of the Primary Processes and Mechanism of Photopolymerization, The Journal of Chemical Physics, 40(1), 1992-1997. [ Links ]
12. Doepker, R. 1968, Vacuum-Ultraviolet Photolysis of the C4H6 Isomers. 1,3-butadiene, The Journal of Physical Chemistry, 72 (12), 4037-4042. [ Links ]
13. Liu, X., Jeffries, E., Sexton, K. 1999, Hydroxyl radical and ozone initiated photochemical reactions of 1,3-butadiene, Atmospheric Environment, 33, 3005-3022. [ Links ]
14. Jaoui, M., Lewandowski, M., Docherty, K., Offenberg, J., Kleindienst, T. 2014, Atmospheric oxidation of 1,3-butadiene: Characterization of gas and aerosol reaction producís and implications for PM2.5, Atmos. Chem. Phys., 14, 13681-13704. [ Links ]
15. Schomaker, V., Pauling, L. 1939, The Electron Diffraction Investigation of the Structure of Benzene, Pyridine, Pyrazine, Butadiene-1,3, Cyclopentadiene, Furan, Pyrrole, and Thiophene, J. Am. Chem. Soc, 61, 1769-1780. [ Links ]
16. Almenningen, A., Bastiansen, O., Traetteberg, M. 1958, An Electron Diffraction Reinvestigation of the Molecular Structure of 1,3-Butadiene, Acta Chemica Scandinavica, 12, 1221-1225. [ Links ]
17. Radom, L., Pople, J. 1970, Rotational Barriers of Alkyl Cations, Journal of the American Chemical Society, 92, 6380-6383. [ Links ]
18. Skaarup, S., Boggs, J., Skancke, P. 1976, Contributions of resonance, hybridization, and nonbonded interactions to the structure of butadiene, Tetrahedron, 32, 1179-1181. [ Links ]
19. Bock, Ch., Panchenko, Y. 1989, An ab initio structural investigation of 1,3-butadiene, isoprene and 2,3-dimethyl-1,3-butadiene rotamers, Journal of Molecular Structure (Theochem), 187, 69-82. [ Links ]
20. Murcko, M. A., Castejon, H., Wiberg, K. B. 2006, Carbon-Carbon Rotational Barriers in Butane, 1-Butene, and 1,3-Butadiene, J. Phys. Chem., 100, 16162-16168 [ Links ]
21. Sancho-García, J., Perez-Jimenez, A., Moscardo, F. 2001, Description of C(sp2)-C(sp2) Rotation in Butadiene by Density Functionals, 705(51), 11541-11548. [ Links ]
22. Feller, D., Craig, N. 2009, High Level ab Initio Energies and Structures for the Rotamers of 1,3-Butadiene, J. Phys. Chem. A., 113, 1601-1607. [ Links ]
23. Atkinson, R., Perry, R., Pitts, J. 1977, Absolute rate constants for the reaction of OH radicals with aliene, l,3butadiene, and 3 methyllbutene over the temperature range 299-424°K, The Journal of Chemical Physics, 67(7), 3170-3175. [ Links ]
24. Fantechi, G., Jensen, N., Saastad, O., Hjorth, J., Peeters, J. 1998, Reactions of Cl atoms with selected VOCs: Kinetics, producís and mechanisms, Journal of Atmospheric Chemistry., 31, 247-267. [ Links ]
25. Francisco-Márquez, M., Alvarez-Idaboy, J., Galeano, A., Viever-Bunge, A. 2005, A Posible Mechanism for Furan Formation in the Tropospheric Oxidation of Dienes, Environ. Sci. Technol, 39, 8797-8802. [ Links ]
26. Li, Z., Nguyen, P., Leon, M., Wang, J., Han, K., He, G. 2006, Experimental and Theoretical Study of Reaction of OH with 1,3-Butadiene, J. Phys. Chem. A., 110, 2698-2708. [ Links ]
27. Ghosh, B., Bugarin, A., Connell, B., North, S. 2010, OH radical initiated oxidation of 1,3-butadiene: Isomeric Selective Study of the Dominant Addition Channel, J. Phys. Chem. A., 114, 5299-5305. [ Links ]
28. Liu, A., Mulac, W., Jonah, C. 1988, Rate Constants for the Gas-Phase Reactions of OH Radicals with 1,3-Butadiene and Aliene at 1 atm in Ar and over the Temperature Range 305-1173 K. J. Phys. Chem., 92, 131-134. [ Links ]
29. Liu, A., Jonah, C, Mulac, W., 1989, The Gas-Phase Reactions of Hydroxyl Radicals with several Unsaturated Hydrocarbons at Atmosphere Pressure, Radial Phys. Chem., 34 (4), 687-691. [ Links ]
30. Pople, J. A., Head-Gordon, M., & Raghavachari, K. 1987, Quadratic configuration interaction. A general technique for determining electrón correlation energies, The Journal of Chemical Physics, 87(10), 5968-5975. [ Links ]
31. Becke, A.D. 1997, Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals, J. Chem. Phys., 107, 8554-8560. [ Links ]
32. Lee, C, Yang, W., & Parr, R. G. 1988, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Physical review B., 37(2), 785. [ Links ]
33. Dunning, T.H. 1971, Gaussian Basis Functions for Use in Molecular Calculations. III. Contraction of (10s6p) Atomic Basis Sets for the First Row Atoms, J. Chem. Phys., 55, 716-723. [ Links ]
34. Frisch, M., Trucks, G., Schlegel, H., Scuseria, G., Rob, M., Cheeseman, J., Montgomery, J., Vreven, T., Kudin, K., Burant, X., Millam, X., Iyengar, S., Tomasi, X.,Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, X., Ishida, M.,Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, X., Hratchian, H., Cross, X., Bakken, V., Adamo, C, Xaramillo, X., Gomperts, R., Stratmann, R., Yazyev, O., Austin, A., Cammi, R., Pomelli, C, Ochterski, X., Ayala, P., Morokuma, K., Voth, G., Salvador, P., Dannenberg, X., Zakrzewski, V., Dapprich, S., Daniels, A., Strain, M., Farkas, O., Malick, D., Rabuck, A., Raghavachari, K., Foresman, X.,Ortiz, X., Cui, Q., Baboul, A., Clifford, S., Cioslowski, X., Stefanov, B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R., Fox, D., Keith, T., Al-Laham, M., Peng, C, Nanayakkara, A., Challacombe, M., Gilí, P., Xohnson, B., Chen, W., Wong, M., González, C, y Pople, X. 2003, Gaussian 03 (Gaussian, Inc., Wallingford, CT), https://gaussian.com/g03citation/. acces date: 05/20/2019. [ Links ]
35. Rodríguez-Linares, D., Freitas, G., Ballester, M., Nascimento, Garrido, X. 2015, Coupled-Cluster Study of the Lower Energy Región of the Ground Electronic State of the HSO2 Potential Energy Surface, J. Phys. Chem. A., 119 (32), 8734-8743. [ Links ]
36. Peterson, K.; Woon, D.; Dunning, T.; Benchamark, X. 1994, Calculationes with correlated molecular wave functions. IV. The classical barrier height of the H+H2H2+H reaction, J. Chem. Phys. 100, 7410-7415. [ Links ]
37. Vasilyev, V. 2017, Online complete basis set limit extrapolation calculator, Computational and Theoretical Chemistry, 1115, 1-3. [ Links ]
38. Engel, T., Reid, P., Hehre, W. Física Química, Ed. Pearson, 2006, Madrid, España, pp. 1090. [ Links ]
39. Parker, C, Coosky, A. 1998, Ab Initio study of the 1,3-butadienyl radical isomers, J. Phys. Chem. A., 102 (30), 6186-6190. [ Links ]

